糖尿病肾病内皮细胞损伤机制的研究进展
2020-10-14陈碧霞张利吴玲玲陈香美
陈碧霞,张利,吴玲玲,陈香美
广东药科大学临床医学院/国家肾脏病临床医学研究中心,北京 100017
糖尿病肾病(diabetic nephropathy,DN)是糖尿病患者的微血管并发症之一,疾病进展的终点为终末期肾病[1]。早期DN肾小球处于高滤过状态,以微量蛋白尿为主,疾病进展后期出现大量蛋白尿,伴随进行性的肾功能下降及高血压[2],病理表现为肾小球改变(包括基底膜增厚、足细胞足突丢失、内皮窗孔丢失及系膜基质增宽等)、肾小球硬化及间质纤维化。近年来研究证实,广泛的内皮功能障碍对肾小球疾病的发生、发展有着重要影响[3-5],肾小球内皮细胞(glomerular endothelial cells,GECs)功能障碍是DN的始动环节,且GECs通过与系膜细胞及足细胞的交流进一步促进了DN的发展[6-7]。
肾脏是一个高度血管化的器官,存在不同种类的内皮细胞,包括中大血管内皮细胞(负责运输血液、调节血管通透性)、肾小管周围毛细血管内皮细胞(参与肾小管分泌和重吸收)及GECs(参与构成肾小球滤过屏障)等。其中,GECs由丝状糖萼和窗孔组成,糖萼有助于调节血管通透性及血流平衡,并排斥血细胞远离血管壁[8],异常丰富的窗孔在水、小溶质通过毛细血管壁的过滤过程中发挥关键作用[9]。内皮功能障碍可引起蛋白尿、肾小球硬化及肾间质纤维化[10],导致肾脏进一步受损,加速DN的进展。因此,本文针对导致肾小球内皮功能障碍的糖代谢紊乱、氧化应激、异常血管生成及炎症等相关机制的研究进展进行了综述。
1 糖代谢紊乱
糖尿病患者存在高血糖症及葡萄糖转运蛋白1(Glut-1)的表达增加,进入细胞的葡萄糖增多,肌细胞可自动调节对葡萄糖的吸收,但内皮细胞则无类似的自动调节作用,因此,高血糖症增加了内皮细胞中葡萄糖及其代谢产物的积累。高血糖通过多元醇途径使葡萄糖代谢增加,细胞内山梨糖醇累积,升高的山梨糖醇水平使超氧化物的产生增加,局部激活肾素-血管紧张素-醛固酮系统(RAAS)[11-12],使血管紧张素Ⅱ(angiotensin Ⅱ,Ang-Ⅱ)生成增多,糖基化终产物受体(receptor of advanced glycation endproducts,RAGE)表达上调,而糖基化终产物(advanced glycation end products,AGEs)与RAGE结合后通过激活NAD(P)H氧化酶(NOX2)[13]增加内皮细胞(endothelial cells,ECs)中活性氧(reactive oxygen species,ROS)的合成,并通过激活磷脂酶C(PLC)诱导二酰基甘油(DAG)的生成,从而导致蛋白激酶C(protein kinase C,PKC)激活。PKC激活后进而激活丝裂原活化蛋白激酶(mitogen activated protein kinase,MAPK)及转录因子如核因子-κB(NF-kappa B,NF-κB)和激活蛋白1(activator protein 1,AP-1),PKC介导的NF-κB激活可使足细胞合成乙酰肝素酶增加,从而促进内皮细胞糖萼的破坏[10]。同时,增多的DAG可使PKC βⅡ/JNK/Bcl-2途径被激活,导致内皮细胞凋亡[14]。
2 氧化应激
氧化应激是糖尿病及肥胖相关肾病等代谢性疾病出现血管并发症的重要致病因素[15-16]。一氧化氮(nitric oxide,NO)与ROS生成之间的不平衡是导致糖尿病内皮功能障碍的主要机制。研究证实,DN小鼠中肾小球氧化应激的增强主要集中在GECs[17-19], 氧化应激是内皮细胞功能障碍的主要机制[20]。糖尿病导致肾小球中NADPH氧化酶(Nox4)及转化生长因子-β1(transforming growth factor β1,TGF-β1)的表达上调,从而促进ROS的产生[21]。ROS过度累积可将四氢生物蝶呤(tetrahydrobiopterin,BH4)氧化为非活性代谢物,促进内皮型一氧化氮合酶(endothelial nitric oxide synthase,eNOS)解耦反应,导致内皮细胞中NO的产生减少[22]。同时,ROS升高还会导致线粒体稳态失调及线粒体膜电位去极化,而线粒体功能障碍可促进释放细胞色素C,通过激活胱天蛋白酶(caspases)及P53蛋白的表达触发凋亡途径,最终导致细胞死亡[23-24]。增加的ROS促进了AGEs的形成,通过激活NADPH-氧化酶途径与受体RAGE结合,并进一步增加内皮细胞中ROS的合成[13]。在易感DN的小鼠及DN患者中发现,GECs线粒体ROS的过度生成可导致内皮细胞损伤,这与肾小球内皮素1受体(endothelin 1 receptor,EDNRA)的表达增加有关[25]。通过清除线粒体ROS或选择性阻断EDNRA可减轻糖尿病引起的内皮损伤、蛋白尿、足细胞丢失及肾小球硬化,从而改善内皮细胞线粒体的氧化应激。
3 异常的血管生成
早期DN存在异常的血管生成,可致肾小球肥大及微量蛋白尿。轻度损伤的GECs主要增加了血管内皮生长因子受体(vascular endothelial growth factor receptor,VEGFR)的激活,血管内皮生长因子(vascular endothelial growth factor,VEGF)与VEGFR2结合发生自身磷酸化,并通过PI3K/Akt信号途径使细胞内钙离子水平升高,钙离子与钙调蛋白结合后激活eNOS表达,导致NO产生增加。VEGF信号传导还可使eNOS mRNA及蛋白质水平升高,eNOS表达增加,促进血管通透性增高及内皮细胞的存活[26]。此外,VEGF/VEGFR2信号还可激活磷脂酶γ(phospholipase gamma,PLC γ),使PKC表达增加,前列腺素2(prostaglandin 2,PGI2)水平升高[26],从而促进血管新生。富亮氨酸α2糖蛋白1(leucine-rich alpha-2-glycoprotein 1,LRG1)是一种促血管生成因子,主要表达于GECs中,研究证实,肾小球LRG1参与了异常血管的生成,其机制独立于VEGF信号传导途径[27]。高糖及炎症导致GECs中LRG1表达升高,通过募集TGF-β辅助受体ENG促进TGF-β与激活素受体样激酶1(activin receptor like kinase 1,ALK1)受体结合,磷酸化Smad1/5/8,导致内皮细胞异常血管生成。LRG1基因缺失可明显减轻早期DN动物模型的肾功能损伤及异常血管生成[28]。轴突导向因子(Netrin-1)可与Unc-5 Netrin受体B(UNC5B)结合调节血管生成。Jiao等[29]在DN患者及链脲佐菌素诱导的DN大鼠GECs中发现,UNC5B表达上调,并与Netrin-1竞争性结合后可抑制VEGR下游蛋白酪氨酸激酶(Src protein tyrosine kinase,SRC)信号传导,最终导致异常的血管生成;通过去除内源性UNC5B并增加外源性Netrin-1可抑制肾小球的异常血管生成,降低肾小球滤过率及蛋白尿。神经迁移蛋白(Slit2)是一种分泌蛋白,高糖环境下,内皮细胞Slit2/Robo1信号激活并参与异常血管的生成,阻断该途径可以抑制异常血管生成的过程[30]。综上所述,LRG1、Netrin-1/UNC5B及Slit2/Robo1是治疗早期DN异常血管生成的潜在靶点。
4 炎症反应
炎症是导致DN的重要病理生理基础。高血糖症导致AGEs增多,通过RAGE-NF-κB信号通路诱导巨噬细胞迁移并释放炎症因子TNF-α及IL-1β[31],促进内皮细胞黏附分子的表达,同时减少保护分子如血栓调节蛋白的产生[32]。炎性因子还能诱导内皮细胞糖萼脱落,从而增加黏附分子的暴露及白细胞的募集,加速DN的进展[10]。趋化素(Chemerin)是一种脂肪细胞因子,可刺激炎性因子释放并诱导炎性细胞浸润,趋化素通过与其受体趋化素受体23(ChemR23)结合,增加TGF-β1及其他炎性因子的表达而导致内皮细胞炎症,加重DN进程[33]。蛋白激酶受体1-4(PAR1-4)是表达于内皮细胞表面的G蛋白偶联受体(GPCRs),被凝血蛋白酶特异性激活后可调节内皮细胞的炎症反应。研究发现,糖尿病患者中存在凝血功能障碍,凝血酶激活PAR1后上调内皮细胞的炎性细胞因子如IL-1、IL-6、TNF以及细胞黏附分子如E-选择素、细胞间黏附分子-1(intercellular adhesion molecule 1,ICAM-1)、血管细胞黏附分子-1(vascular cell adhesion molecule 1,VCAM-1)等的表达,促进内皮细胞凋亡,增加其通透性[34],而大量细胞凋亡可引发炎症反应[35]。Gasdermin D(GSDMD)是一种新发现的重要的细胞凋亡执行蛋白,可被caspases裂解,高糖可诱导内皮细胞中Nod样受体蛋白3(NLRP3)炎性小体活化,活化的NLRP3通过caspase-1把GSDMD裂解为蛋白水解片段,在细胞膜上形成孔并导致细胞死亡,释放大量促炎因子如IL-1β[36],增加黏附因子ICAM-1及VCAM-1的表达,导致内皮细胞炎症的发生[37]。Anagliptin是一种二肽基肽酶4(DPP4)抑制剂,可通过上调沉默信息调节因子1(silent information regulator 1,SIRT1)表达及激活Nox4-ROS-TXNIP通路抑制高糖诱导的内皮细胞NLRP3活化及caspase-1表达,对内皮细胞起保护作用[38]。
5 内皮间充质转化(endothelial-mesenchymal transition,EndMT)
肾纤维化是DN发展至晚期的一个重要标志,EndMT是内皮功能障碍的特殊特征,可致微血管稀疏[39],并促进DN肾纤维化的进展[40]。内皮细胞谱系示踪显示,在链脲佐菌素诱导的DN小鼠纤维化肾间质中存在内皮来源的α-平滑肌肌动蛋白(α-SMA)阳性肌成纤维细胞,表明EndMT参与了肾纤维化[41]。Peng等[42]证实在DN患者及db/db糖尿病小鼠的GECs中发生了EndMT。异常的TGF-β信号传导具有促进纤维化作用,TGF-β1/Smad信号通路是导致EndMT的最主要通路之一[43]。高血糖症及其相关代谢和血流动力学改变可使糖尿病动物模型及DN患者分泌TGF-β,TGF-β活化激活素受体样激酶5(activin receptor like kinase 5,ALK5)受体,导致Smad2/3磷酸化[44],并与Smad4形成异源二聚体易位至细胞核,直接与DNA相互作用,以调节TGF-β靶基因的转录[45]。Li等[46]在链脲佐菌素诱导的DN小鼠中证实了TGF-β/Smad3信号通路促进EndMT的过程。Zhao等[47]在糖尿病动物模型GECs中发现刺猬相互作用蛋白(Hhip)表达升高,随后激活TGF-β1/Smad级联反应促进EndMT,最终导致纤维化。然而,Saura等[48]发现,TGF-β1/Smad信号通路的激活也会诱导内皮细胞中eNOS表达上调,可能对内皮细胞具有保护作用。TGF-β信号传导对内皮细胞的作用仍需更多的实验进行探究。除TGF-β外,AGEs可通过与RAGE受体结合诱导内皮细胞中的Smad3激活,促进EndMT的进程,而使用Smad3抑制剂SIS3可降低EndMT,延缓DN的发展[46]。 氧化应激可促进EndMT过程,Lin等[49]通过体外实验验证了氧化应激通过激活SIRT3-Foxo3a-过氧化氢酶信号通路导致EndMT及肾纤维化。除TGFβ1-Smad2/3及SIRT3-Foxo3a-过氧化氢酶通路外,DKK3-Wnt通路在EndMT及肾纤维化中也有作用。研究发现,Dickkopf相关蛋白3(DKK3)可抑制肾脏内皮细胞的生长、促进肌成纤维细胞的形成,并激活EndMT,而Wnt通路抑制剂可下调DKK3的表达,同时抑制肾脏纤维化[50]。高糖可导致Rho相关激酶(Rho-associated kinase,ROCK1)表达升高,诱导内皮细胞EndMT的发生,使用ROCK1抑制剂可阻断EndMT,同时阻止蛋白尿的进展[42]。
6 自 噬
自噬对内皮细胞的调节具有重要作用。自噬可调节NO的产生、血管生成及血栓形成,内皮细胞自噬受损可改变细胞结构,导致内皮细胞向间充质转化[51]。近年研究发现,自噬参与了DN内皮细胞的损伤,高糖通过抑制腺苷酸活化蛋白激酶(adenosine monophosphate activated protein kinase,AMPK)依赖的LKB1/STRAD/MO25复合物形成,降低了G E C s 自噬标志物如自噬基因相关蛋白5(autophagy gene-associated protein 5,Atg5)、Atg7、Atg3的表达,从而降低微管相关蛋白质1轻链3 B/A (microtubule-associated protein 1 light chain 3 B/A,LC3B/A)的比值,抑制自噬的发生[52]。体外实验中特异性敲除内皮细胞的Atg5可导致糖尿病引起的GECs损伤[53],而在GECs的Atg5敲除小鼠中出现肾小球形态异常及ROS积累,提示内皮细胞的自噬可减轻肾小球氧化应激损伤并能维持肾小球毛细血管的完整性[54],表明自噬对内皮细胞及DN具有保护作用。
7 内皮细胞与其他细胞间的通讯
内皮细胞损伤在早期DN的发生、发展中起关键作用,它与肾小球足细胞、系膜细胞及肾小管上皮细胞在解剖位置上可能存在相互串扰,并影响DN的发展,因此,研究内皮细胞与其他细胞间的串扰具有十分重要的意义。
7.1 内皮细胞与足细胞 足细胞通过与内皮细胞串扰来维持毛细血管内环境稳态[55],足细胞与内皮细胞损伤共同参与了肾小球疾病的进展。血管内皮生长因子A(vascular Endothelial growth factor receptor,VEGF-A)及其受体VEGFR系统对正常的肾小球发育及维持肾脏稳态至关重要,其变化在DN进展中起主要作用。足细胞合成VEGF,通过旁分泌的方式与表达在内皮细胞表面的VEGFR结合发挥作用。DN早期,各种因素导致内皮细胞的VEGFR表达升高,足细胞可通过维持较高水平的基础自噬以快速响应由内皮细胞引起的细胞毒性[56],使VEGF/VEGFR表达上调从而促进肾小球血管新生。然而到DN晚期,VEGF/VEGFR表达下调则导致血管稀疏及肾脏纤维化。其原因可能是到疾病后期,损伤的内皮细胞通过胞外囊泡影响足细胞的细胞骨架排列、线粒体功能并增加miRNA-200c-3p的表达[57], 足细胞自噬不足[56]导致自身损伤严重,VEGF/VEGFR产生减少。血管紧张素/血管生成素受体2 (Ang/Tie-2)系统在维持内皮完整性中起至关重要的作用,并参与了DN的病理生理过程。Ang-Ⅰ主要在足细胞中表达,调节VEGF水平,促进细胞存活并维持正常内皮细胞功能[58]。Ang-Ⅱ是内皮细胞中Tie-2的拮抗配体,以自分泌方式抑制Ang-Ⅰ与Tie-2结合。高糖及AGEs通过降低Ang-Ⅰ诱导的Akt及叉头框转录因子O1(forkhead box transcription factor O-1,FoxO1)磷酸化,并增加了Ang-Ⅱ的表达,抑制内皮细胞增殖、增加ROS产生并降低氧化型及还原型谷胱甘肽间的比值,导致内皮细胞功能障碍[59]。 内皮素1(endothelin 1,ET-1)是一种具有促炎及促纤维化作用的血管收缩肽,主要由GECs表达。足细胞通过激活TGF-β R1信号激活相邻内皮细胞的内皮素A受体(ETAR),抑制ETAR可预防足细胞丢失、白蛋白尿及肾小球硬化[17]。此外,损伤的内皮细胞还可通过外泌体的作用导致EndMT及足细胞功能障碍[60]。
7.2 内皮细胞与系膜细胞 肾小球硬化是DN的主要病理特征,肾小球基底膜增厚、细胞外基质(extracellular matrix,ECM)增多是肾小球系膜细胞表型发生改变所致。GECs可与系膜细胞直接接触并相互作用,如GECs分泌的血小板衍生生长因子B(platelet-derived growth factor B,PDGFB)与系膜细胞上的血小板衍生生长因子受体β(platelet-derived growth factor receptors β,PDGFR-β)结合有助于系膜细胞发育。沉默PDGFB基因及其受体PDGFR-β基因可减少系膜细胞的增殖及ECM的累积[61]。在糖尿病中,损伤的GECs通过旁分泌机制导致系膜细胞损伤,促进DN的进展[62],其机制是高糖通过激活PI3K/Akt及MAPK通路介导内皮细胞分泌更多富含circRNA的外泌体,促进了α-SMA的表达,并通过TGF-β1/Smad3信号通路影响系膜细胞的增殖并促进EndMT,最终导致肾纤维化[63]。另外有研究发现,内皮细胞表达的ET-1介导了肾小球系膜细胞中ETAR的活化,使RhoA/ROCK通路被激活,导致了系膜细胞的增殖及ECM的累积[64]。
7.3 内皮细胞与肾小管上皮细胞 DN早期有不同程度的肾小管损伤,肾小管上皮细胞及GECs间的串扰在DN的发生和发展中起着重要作用[65-66]。肾小管上皮细胞损伤可导致内皮细胞功能出现异常。氧化应激可通过NF-κB及其他信号通路介导GECs损伤、凋亡及EndMT的发生[67]。DPP-4抑制剂作用于远端肾小管时,可减轻肾脏组织的氧化应激水平,防止炎性因子引起的内皮细胞损伤,并减少蛋白尿排泄[68]。此外,肾小管上皮细胞可通过Ang-Ⅰ/Ang-Ⅱ-Tie-2轴[67]及VEGF/VEGFR轴[69]调节内皮细胞功能。同时,受损的内皮细胞也可通过产生Kruppel样因子(KLF)[70-71]、肝细胞生长因子(HGF)[72]、 胰岛素样生长因子结合蛋白(IGFBP)[67,73]反过来影响肾小管上皮细胞的形态及功能。如何维持内皮细胞与其他细胞间正常的串扰关系可能成为预防和治疗DN的新策略。
8 表观遗传学
长期处于高糖状态下的DN患者存在代谢记忆现象,即去除原始刺激因素后,代谢记忆仍会导致糖尿病并发症的发生[74]。因此,有学者提出表观遗传学可能与DN的进展有关[75]。在DN患者肾脏中发现存在显著差异的DNA甲基化基因,为表观遗传学与DN间的关系提供了直接证据[76]。高糖可诱导表观遗传的改变,如关键基因启动子区高度甲基化及组蛋白修饰,从而使糖基化产物及自由活性氧等代谢产物累积[77],调控下游NF-κB、RAGE及纤溶酶原激活剂抑制剂-1(plasminogen activator inhibitor-1,PAI-1) 等基因的表达,进而引起病理性血管新生、血管通透性改变及抑制DNA合成等病理性过程[78-79]。此外,有研究发现,miRNA参与了调节DN的进展,如在糖尿病db/db小鼠GECs中发现miRNA-93下 调[80],miR-29c、miR-129及miR-200b上调[81],共同影响血管生成,而miR-146a下调促进了氧化应激及肾功能障碍[74]。上述证据表明,表观遗传机制通过微调内皮细胞基因的表达促进疾病进展,并可能是代谢记忆现象的关键机制之一,进一步深入研究内皮细胞关键基因的表观遗传机制及相关信号转导途径,有助于深入阐明DN的发生发展机制,而消除代谢记忆为研发有效的治疗药物提供了新的靶点[79]。
9 外泌体
外泌体是从细胞分泌到细胞外空间的囊样体,在肾脏中具有维持细胞间通讯及信号传导、参与免疫调节的作用[82]。在糖尿病患者的血液、尿液中已检测到可作为潜在临床诊断标志物的各种外泌体[83], 而内皮细胞损伤在DN早期出现,内皮细胞已被证明可以分泌外泌体,并促进DN的进程[83-85],因此,研究内皮细胞外泌体对于早期诊断和防治DN具有重要意义。高糖状态下,内皮细胞产生富含TGF-β1mRNA的外泌体,导致上皮间充质转分化(epithelial-mesenchymal transition,EMT)及肾小球系膜细胞表达α-SMA[63],促进纤维化[60,86];通过分泌含有circRNA-0077930[87]、Versican蛋白[88]及Notch3[87]等的外泌体诱导血管平滑肌细胞钙化或衰老。外泌体除传递损伤信号外,还具有保护作用,如内皮集落形成细胞(ECFC)[89]及内皮祖细胞(EPC)[90]分泌的外泌体具有抑制内皮细胞凋亡,促进增殖、迁移的作用。缺氧状态下,内皮细胞释放的外泌体通过上调赖氨酰氧化酶样蛋白2增加胶原蛋白交联活性,促进内皮细胞修复[91]。综上,除挖掘具有临床诊断意义的外泌体外,还可根据外泌体的不同作用研发治疗DN的新型药物。
10 总结与展望
GECs以其独特的结构和功能在肾小球滤过屏障正常功能的发挥中起着关键作用。GECs损伤的机制可能是通过糖代谢紊乱、氧化应激、炎症反应、异常血管生成等所致(图1)。近年来研究表明,肾脏细胞间存在串扰,作为终末分化的细胞,内皮细胞极易受到其他损伤细胞的影响而发生结构或功能的改变,如损伤的肾小管上皮细胞及足细胞可使GECs炎症及氧化应激水平增高,导致其凋亡及病理性的血管增生[62,67]。此外,各种研究发现,当内皮细胞表观遗传学发生改变时,其损伤程序不会因为去除致病因素而终止,由于代谢记忆的存在,内皮细胞持续表达致病基因,使自身及周围其他细胞发生损伤,最终导致肾脏细胞外基质增多,表现为肾功能不全。
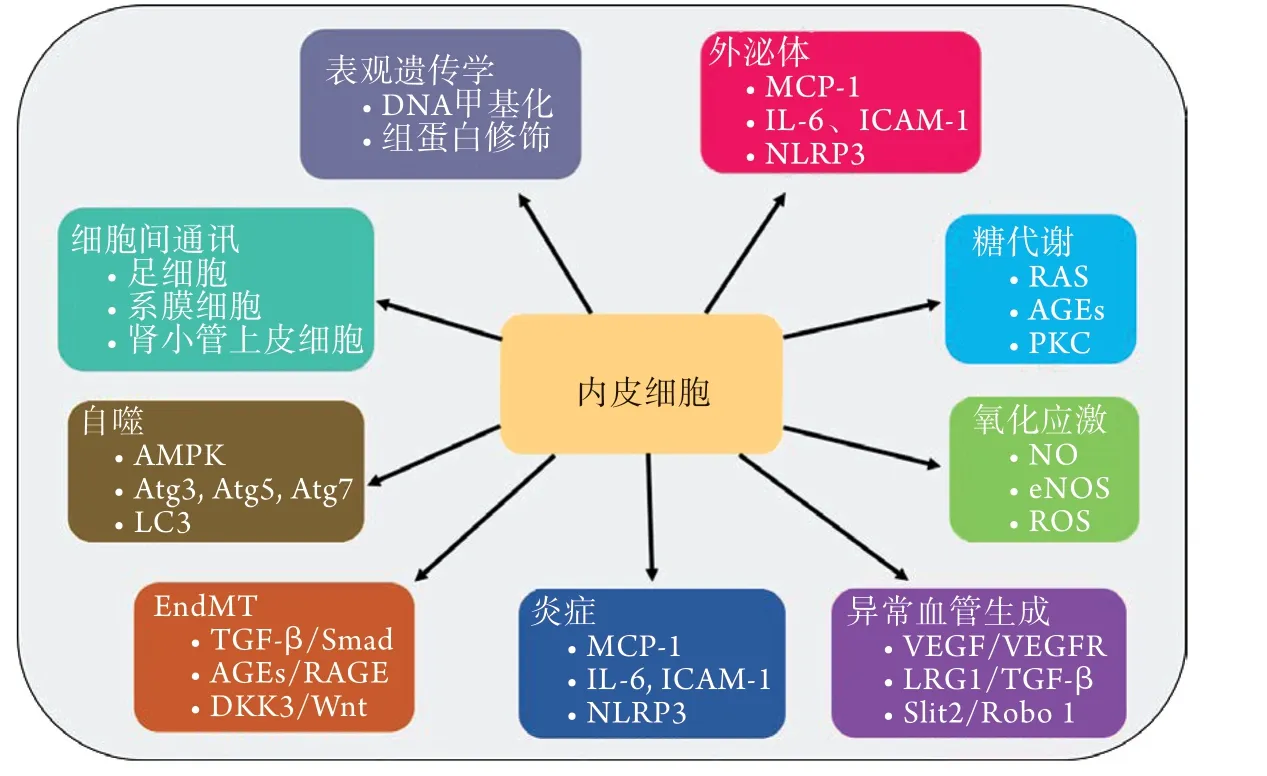
图1 糖尿病肾病内皮细胞损伤机制示意图Fig.1 The mechanism of endothelial cells injury in diabetic nephropathy
传统的抗氧化、抗纤维化、抗VEGF及保护糖萼等措施可缓解DN的发展,但对疾病中晚期治疗效果不明显,疾病仍向终末期肾病发展。最近几年,临床前模型及临床研究发现,钠糖共转运蛋白2(sodium-sugar co-transporter 2,GLT2)抑制剂可减轻系统性大血管和肾脏微血管僵硬度,减缓DN进程,其机制可能与抑制AGEs的生成、恢复eNOS活性、减少和降低血管炎症信号传导有关[92-94]。SGLT2抑制剂对内皮细胞的影响具有特异性,如卡格列净可抑制人类内皮细胞增殖及血管形成,但依帕格列净或达格列净则未发现这类作用。关于SGLT2抑制剂对内皮细胞的作用机制及具体信号通路仍需大规模临床试验的验证。
治疗DN的关键在于早期诊断和预防。目前对早期DN仍无精准的诊断方法,而作为在DN早期即出现损伤的GECs,探究其损伤标志物及如何将检测GECs损伤的方法用于DN的早期诊断及防治,是未来的研究方向。
