五元环氟化物的合成及应用
2020-09-29张呈平庆飞要贾晓卿权恒道
张呈平,庆飞要,贾晓卿,权恒道
(北京理工大学化学与化工学院,北京100081)
引 言
大气臭氧空洞和全球暖化是当今国际社会面临的两大环境难题。研究认为,氟利昂(氟氯烃,CFCs)是破坏大气臭氧层的主要元凶;温室气体的大量排放,加剧了温室效应,是造成全球暖化的主要原因。为保护大气臭氧层及减少温室气体排放,国际社会在1987 年和2016 年分别签署了限制使用CFCs 的《蒙特利尔议定书》和减排或限制使用氢氟氯烃(HCFCs)、氢氟烃(HFCs)、SF6等温室气体的《巴黎协定》。因此,针对CFCs替代这一世界的重大需求,科学家进行了长期的努力,先后开发出了四代CFCs 替代物:第一代是氢氟氯烃(HCFCs),臭氧损耗潜值(ODP)比CFCs 低,全球暖化潜值(GWP值)很高,温室效应显著,属于过渡品种;第二代是氢氟烃(HFCs)和氢氟醚(HFEs),其ODP 值为零,但GWP 值很高,大多属于高温室气体,现在处于逐步削减的阶段;第三代是氢氟烯烃(HFOs)和环状氟化物,其ODP 值为零,GWP 值较低(GWP 值<250),处于推广和应用的阶段;第四代是杂原子氟化物(杂原子可以是N、O、S 等),其ODP 值为零,GWP 值很低,大多属于高附加值的精细氟化工品,如七氟异丁腈[1−3]、碳酰氟[4]、三氟碘甲烷[5]、三氟甲基全氟异丙基酮[6]等。
在第三代CFCs 替代物的开发中,顺式或反式−1,3,3,3−四氟丙烯[HFO−1234ze(Z)或HFO−1234ze(E)],以2,3,3,3−四氟丙烯(HFO−1234yf)、顺式−1,1,1,4,4,4−六 氟−2−丁 烯[HFO−1336mzz(Z)]为 代 表 的HFOs 的合成专利技术基本被杜邦、霍尼韦尔、新化学品公司所垄断。国内有关HFO 类物质的合成方法和合成工艺及应用研究,与国外大公司相比差距较大。
五元环氟化物是重要的环状氟化物。与三元环、四元环相比,由于五元环中碳−碳键的张力更小,其碳−碳键的夹角更接近四面体杂化的夹角,因此,五元环的稳定性更强。另外,氟原子引入五元环化合物,氟原子会降低ODP 值甚至为零,也会增加化合物的流动性、传热性能和稳定性,同时也会稍微增加GWP 值,使五元环氟化物具有特殊的性质,应用于特定领域。例如:1,1,2,2,3,3,4−七氟环戊烷(F7A)、1,1,2,2,3,3−六氟环戊烷(F6A)、顺式−1,1,2,2,3,3,4,5−八氟环戊烷(cis−F8A)、八氟环戊烯(F8E)、1,3,3,4,4,5,5−七氟环戊烯(F7E)和3,3,4,4,5,5−六氟环戊烯(F6E)等五元环氟化物,已经被确认属于环境友好型物质[7−10],在电子清洗[11]、电子刻蚀[12−13]、高温热泵[14]等领域具有重要的应用。然而,五元环氟化物的碳原子数比一般的C3和C4的HFOs更高,反应过程更复杂,合成难度更高,技术门槛较高,导致国际上在五元环氟化物领域游刃有余的院校、科研院所和企业屈指可数。经过多年开发,我国建成了世界首套工业化联产五元环氟化物的生产线。
本文就1,2−二氯六氟环戊烯(F6−12)、1,3−二氯六氟环戊烯(F6−13)、1−氯七氟环戊烯(F7−1)、F7A、F6A、cis−F8A、F8E、F7E 和F6E 等五元环氟化物的合成进行综述(图1),并对五元环氟化物在电子清洗、电子刻蚀、合成电子氟化液等领域的应用进展进行跟踪,以期对第三代CFCs替代物的深入开发与产业化具有一定的指导作用。
1 五元环氟化物的合成
1.1 F6-12或F6-13的合成
F6−12、F6−13是合成系列五元环氟化物的重要中间体。其中,F6−12 可通过六氯环戊二烯(HCCPD)或1,2,3,4−四氯环戊烷(TCCPA)的氯氟化反应或八氯环戊烯(OCP)的氟化反应合成得到,而F6−13 可通过F5−123 或F5−133 的氟化反应合成得到。
F6−12 和F6−13 的传统路线大多采用液相氟化反应工艺。Henne 等[15]报道了OCP 与三氟化锑、氯气发生液相氟化反应,得到F6−12,其产率为50%。Latif[16]报道了OCP 与SbF3和SbF3Cl2的混合物发生液相氟化反应得到F6−12。Michael等[17]报道了在五氯化锑存在下,OCP 与无水氟化氢(HF)发生液相氟化反应,140℃反应7.5 h,得到产率为87.2%的F6−12,其纯度为98.5%。Mcbee 等[18−19]报道了100℃时,HCCPD 与五氟化锑发生液相氟化反应,得到沸点为89~90℃的F6−12,其产率为50%。Henne 等[15]报道了HCCPD 在SbF3Cl2存在下,与HF 和氯气发生液相氯氟化反应,得到F6−12。Zhang 等[20−21]报道了在DMF 溶剂中,1,2,3−三氯五氟环戊烯与KF 发生氟化反应,得到F6−13 和F6−14,其中F6−13 和F6−14 的物质的量之比接近2/1。上述路线存在以下缺陷:(1)属于间歇性工艺,难以实现连续化大规模生产;(2)采用大量溶剂和氟化试剂,在合成过程中产生大量废液和废固,容易污染环境。

图1 系列五元环氟化物的主要合成路线Fig.1 Main synthetic routes of series five−membered ring fluoride
F6−12 也可使用OCP 的气相氟化反应进行制备。先将SbCl5负载到活性炭上得到前体SbCl5/C,该前体在150℃时,先由氮气稀释的HF 活化1 h,再由氮气稀释的氯气活化1 h,制备得到锑基催化剂,然后在该锑基催化剂存在下,OCP 流量28.3 g/h,HF流量44.0 g/h,反应温度175℃,反应时间3.5 h,反应得到的F6−12 产率为77.4%[22]。该方法存在以下缺陷:(1)原料OCP 的熔点较高(40℃),容易堵塞管路,影响连续反应的正常进行;(2)锑基催化剂流失严重,且积炭严重,会快速失活。
为解决上述技术难题,开发了以HCCPD为起始原料,经历气相氯氟化反应,制备F6−12 的合成路线。在铬基催化剂存在下,温度420℃,n(HF)/n(Cl2)/n(HCCPD)=10/1/1,接触时间10 s,连续运行500 h。结果显示,500 h 内,HCCPD 转化率恒定在100%,F6−12选择性约为78%,1,2,4−三氯五氟环戊烯(F5−124)选择性约为13%,F7−1的选择性约为4%[23]。该路线具有原料熔点较低(9.6℃)、原料容易获取、转化率高、选择性好、催化剂使用寿命长的优点。唯一的不足是HCCPD 原料的价格过于昂贵,推高了F6−12的生产成本。
为降低F6−12 的生产成本,开发了DCPD 为起始原料,经历气相热解、液相氯化、气相氯氟化共三步反应,合成了F6−12[24],具体如下:(1)在惰性氧化铝存在下,反应温度350℃,在反应器中同时通入氮气和DCPD,控制n(N2)/n(DCPD)为1/1.5,接触时间为15 s,反应压力0.1 MPa,反应产物经0℃冰浴冷却后,用GC 分析有机物的组成,环戊二烯(CPD)的收率为99.4%;(2)在高压釜中同时加入CPD 和氯气,控制n(CPD)/n(Cl2)为1/1.5,高压釜温度为20℃,反应时间为5 h,产物经水洗、碱洗,然后用4A 分子筛干燥,取样进行GC 检测,TCCPA 的收率为98.2%;(3)铬基催化剂催化TCCPA 的气相氯氟化反应,条件为温度400℃,n(O2)/n(HF)/n(Cl2)/n(TCCPA)为1/20/10/1,接触时间6.86 s,连续运行500 h。结果显示,500 h内,TCCPA 转化率恒定在100%,F6−12 选择性约为80%,F5−124 选择性约为10%,F7−1 的选择性约为5%。该路线具有原料容易获得且价格低廉、转化率高、选择性好、催化剂使用寿命长的优点,容易实现F6−12的连续化大规模生产。
在HCCPD 或TCCPA 的氯氟化反应中,除了得到主要产物F6−12 和少量F7−1 和F8E 外,还可以得到F6−13、F6−14、F5−144、F5−133、F5−123、F5−124、1,2,3,3−四氯四氟环戊烯(F4−1233)和1,2,4,4−四氯四氟环戊烯(F4−1244)、1,2,3,3,4,4−六氯二氟环戊烯(F2−123344)、1,2,3,3,5,5−六氯二氟环戊烯(F2−123355)和1,2,3,3,4,4,5−七氯一氟环戊烯(F1−1233445)等中间体[21,23,25],上述结构均通过GC−MS、核磁氟谱和碳谱进行鉴定,这对工业上实现独立循环工艺产生指导作用,只需将氟化不完全的中间体循环进入反应器即可转化为目标产物,从而推动了五元环氟化物的合成工艺的发展。另外,上述中间体存在很多同分异构体,在一定条件下实现相互转化。在氟离子催化下,F2−123344 与F2−123355 之间的异构化是可逆的;F4−1233 和F4−1244 发生可逆的异构化;而F5−144 与F5−133 之间可发生可逆的异构化反应,F5−123 与F5−124 之间也可发生可逆的异构化,F5−133 向F5−123 的异构化不可逆;F6−14和F6−13之间的异构化是可逆的,而F6−13向F6−12的异构化是不可逆的[26−27]。研究认为,在DMF溶剂中,氟化铵可以提供氟离子使卤代环戊烯发生卤迁移异构化反应,主要符合氟离子的加成/消去机制,反应过程中可能会涉及到碳负离子重排反应,从而实现氟原子在五元环上的迁移。其中,氟原子在C(sp3)碳位之间的迁移属于化学平衡,而氟原子从C(sp2)碳位迁移到C(sp3)碳位不属于化学平衡(图2)[26]。通过化学计算,按照化合物能量大小排序为:F5−144 >F5−133 >F5−123 >F5−124,F6−14 >F6−13 >F6−12[26]。因此,可以通过氟离子或氟化催化剂将F5−144 和F5−133 转化为F5−124 和F5−123,在更高温度下,可以彻底转化为最稳定的构型F5−124;同理,也可采用类似的方法将F6−14 和F6−13 转化为最稳定的构型F6−12[27−28]。
在上述中间体中,F5−133 可发生气相氟化反应得到F6−13。反应条件为:在铬基催化剂存在下,反应温度220℃,n(HF)/n(F5−133)=10,接触时间12 s,连续运行500 h。结果显示,500 h 内,F5−133 转化率恒定在97%~98%,F6−13 选择性约为73%,F6−14的选择性约为0.7%,F6−12 的选择性约为1.5%,其他副产物是1,4,4−三氯五氟环戊烯(F5−144)、1,2,4−三氯五氟环戊烯(F5−124)和1,2,3−三氯五氟环戊烯(F5−123)[23,29]。F5−133 的其他同分异构体也可发生气相氟化反应,其中F5−144 为原料时,主产物是F6−14 和F6−13;F−123 或F5−124 为原料时,主要产物是F6−12,副产物中没有F6−13 或F6−14 生成[23]。上述结果表明,气相氟化反应过程中,HF 与含氯中间体优先发生C(sp3)碳位上的氟化。这是因为气相氟化反应属于Swartz 反应,包括以下三个过程[30−31](图3):(1)含C—Cl 键的氯化物与金属氟化物MFx(M 为金属,x 为自然数)通过形成C—F 键和M—Cl键,得到双卤桥结构的中间体;(2)该中间体经过C—Cl 键和M—F 键的同时断裂,得到含C—F 键的氟化物以及金属氯氟化物MFx−1Cl;(3)MFx−1Cl 经过HF 氟化得到MFx和HCl,实现催化剂的再生。在上述过程中,双卤桥结构的中间体的形成和断裂显得尤为关键。与C(sp3)碳位相比,由于C(sp2)碳位对氯原子的强烈吸引,导致双卤桥结构的中间体中C(sp2)—Cl 键比C(sp3)—Cl 键更难以断裂,从而导致C(sp2)碳位的氟化反应比C(sp3)碳位更难以发生。

图2 氟离子催化卤代环戊烯发生卤迁移的两种途径Fig.2 Two ways of halogen migration in halogenated cyclopentene catalyzed by fluoride ions
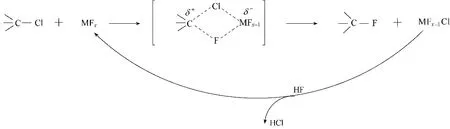
图3 气相氟化反应的机理Fig.3 Mechanism of gas−phase fluorination reaction
1.2 F7-1的合成
F7−1 的传统合成路线大多采用液相氟化方法。Frank[32]报道了F6−12 在DMF 溶剂中与KF 发生液相氟化反应得到F7−1。Mase 等[33]报道了在DMF 与甲苯的混合溶剂中,55.5% F6−12、31.4%三氯五氟环戊烯和12.5%四氯四氟环戊烯组成的氯氟环戊烯混合物与KF在130℃发生液相氟化反应5 h,得到产率为68.8%的F7−1。Yamada 等[34]报道了至少两种氯氟环戊烯的混合物与KF 在极性溶剂与非极性溶剂的混合溶剂中发生液相氟化反应,再将副产物1,3−二氯六氟环戊烯(F6−13)和1,4−二氯六氟环戊烯(F6−14)移除,得到F7−1。
为解决上述难题,开发了气相氟化合成F7−1的合成路线,可以分别以F6−12 或F6−13 为原料,在铬基催化剂存在下,与HF 发生气相氟化反应。其中,F6−12 的气相氟化反应条件为:温度390℃,n(HF)/n(F6−12)=20,接触时间12 s,连续运行500 h。结果显示,500 h 内,F6−12 转化率恒定在35%~40%,F7−1 选择性约为94%,F8E 的选择性约为3%,F5−124选择性约为3%[23,35−37]。该路线存在原料F6−12 容易获得、F7−1选择性好、催化剂使用寿命长、容易实现连续化生产的优点。
另外,F6−13 的气相氟化反应条件为:温度290℃,n(HF)/n(F6−13)=20/1,接触时间12 s,连续运行500 h。结果显示,500 h 内,F6−13 转化率恒定在98%~100%,F7−1 选择性约为78%,F6−12 的选择性约为17%,F6−14 的选择性约为1%,F5−123 的选择性约为1%,F5−124 的选择性约为3%[23,38]。在上述反应中,F6−13 可以被F6−14 替代,同样可以高产率地制备F7−1。该路线具有F7−1 单程产率高、催化剂使用寿命长的特点。但不足之处是该路线的起始原料F5−133 主要来自于TCCPA 或HCCPD 的气相氯氟化的中间体,该中间体属于不稳定构型,在HCCPD 或TCCPA 的气相氯氟化反应中,容易异构化为最稳定构型F5−124,导致原料F5−133 难于获得。
在F6−12和F6−13的气相氟化反应中,F6−13转化为F7−1的产率(约78%)远高于F6−12转化为F7−1 的产率(约40%)。结果表明,F6−13 的3 位—C(sp3) —Cl 的氟化反应比F6−12 的1 位—C(sp2) —Cl更容易发生。其原因是由于C C 双键对氯原子的强吸引,导致在双卤桥结构的中间体中(图3),F6−12 的1 位—C(sp2)—Cl 键比F6−13 的3 位—C(sp3)—Cl键更难以断裂[30−31],也可以解释为F6−13的化合物能量高于F6−12[30],导致F6−13发生氟化反应的能垒低于F6−12。
另外,F7−1 也可以通过1,1−二氯八氟环戊烷(F8−11)的气相加氢脱卤反应得到,催化剂为Cu 改性的Pd 催化剂,反应温度85℃,F8−11 转化率为99.7%,对F7−1 的选择性高达95.8%[39]。其中F8−11可以通过HCCPD、OCP 或F6−12 的氯氟化反应得到,即:在Ta/C、Nb/C 或Sb/C 等催化剂存在下,HCCPD、OCP或F6−12为原料,与HF和Cl2发生反应得到F8−11,在催化剂为Nb/C、反应温度330℃、F6−12 流量48 g/h、HF 流量48 g/h、Cl2流量6 L/h 的条件下,F6−12 转 化 率 为89.2%,F8−11 的 选 择 性 为83.1%[40]。在合成中间体F8−11 的工艺中,采用了活性炭负载Ta、Nb、Sb的氟化催化剂,在高温下发生活性组分流失,另外Ta、Nb、Sb的氟化物属于超强路易斯酸[41],作为活性组分,催化高碳不饱和的有机原料发生氟−氯交换反应,容易积炭而快速失活,并且难以回收再利用。因此,由F8−11 为原料合成F7−1 的加氢脱卤路线存在原料F8−11难以获取的缺陷。
1.3 F8E的合成
F8E的传统合成路线是氯氟环戊烯为原料的液相氟化反应。在DMF 溶剂中,由F6−12、F5−123、1,2,3,5−四氯−3,4,4,5−四氟环戊烯和1,2,3,3,5−五氯−4,4,5−三氟环戊烯按照质量百分比61.7%、30.9%、71.%和0.3%组成的混合物与氟化钾在135℃反应3 h,得到F8E,总收率为93.1%[42]。在DMF 溶剂中,F6−12 与氟化钾在135℃反应0.5 h,得到F8E,收率为87.8%[43]。
为解决上述工艺难题,开发了气相反应合成F8E 的路线。在铬基催化剂存在下,F7−1 与HF 发生气相氟化反应,条件为温度390℃,n(HF)/n(F7−1)=15/1,接触时间12 s,连续反应500 h,则F7−1的转化率恒定在15%~18%,F8E 的选择性约为99%[44]。该路线具有F8E选择性好、催化剂使用寿命长、容易实现连续化生产的优点。
在同等温度条件下,与前面F6−12 的气相氟化反应相比,F7−1的气相氟化反应中原料的转化率更低。其原因是F7−1 中1 位—C(sp2)的电负性(3.198)高于F6−12 中1 位—C(sp2)的电负性(3.121)[45],则F7−1 中1 位—C(sp2)对氯原子的吸引力比F6−12 中1 位—C(sp2)更强,导致在双卤桥结构的中间体(图3)中,F7−1 的1 位C(sp2) —Cl 键比F6−12 的1 位C(sp2)—Cl键更难以断裂[30−31]。
1.4 F7A的合成
F7A 可通过F7E[46]或F8E[47−48]的气相催化加氢反应得到。然而,上述路线均存在原料价格过高的缺陷,而且在F8E加氢脱氟的过程中,对F7A的选择性较低,仅为38%[47−48]。
F7A 也可通过1,1−二氯八氟环戊烷(F8−11)的气相加氢脱卤反应得到,加氢催化剂为Pd+Bi/C、Pd/C、Pt/C 或者Pt+Pd/C,在催化剂为5%Pd+1%Bi/C、反应温度72℃、氢气120 ml/min、F8−11 流量0.2 g/min的条件下,F8−11 转化率为100%,F7A 的选择性为95%[49]。然而该路线存在原料F8−11 难以获得的缺陷。
F7A 还可通过F7−1 的加氢脱氯反应合成得到。Yamada 等[50]研究了在压力反应器中,乙酸钠水溶液为溶剂,催化剂为5%Pd/C,反应压力为980 kPa,F7−1 与氢气发生加氢脱氯反应24 h,得到F7A 和F6A,选择性依次是50%和40%,而反应温度和转化率均没有明确给出;还研究了在装填有催化剂0.5%Pd/Al2O3的管式反应器中,F7−1与氢气发生气相加氢脱氯反应,温度150℃,n(H2)/n(F7−1)=12.7,接触时间1.57 s,连续运行30 h,F7−1转化率约为99%,F7A 选择性约为90.9%。Sekiya 等[51]研究了在温度150℃、n(H2)/n(F7−1)=9.7、接触时间0.1 s 的反应条件下,F7−1 与氢气发生加氢脱氯反应,当催化剂为0.5%Pd/CaF2,2.5 h 时F7−1 的转化率为98.6%,F7A选择性为93.1%,与反应初始相比,转化率下降了1.2%,选择性下降了3.9%;当催化剂为0.5%Pd/C,2.5 h 时F7−1 的 转 化 率 为97.5%,F7A 选 择 性 为84.2%,与反应初始相比,转化率下降了1.6%,选择性下降了4.9%。在上述方法中,液相法存在反应压力较高、选择性较差的不足,气相法则存在催化剂转化率较低、选择性较差、催化剂失活快的缺陷,均不利于工业化生产。
为解决上述技术难题,开发了加氢催化剂Pd+Bi/AlF3用于催化F7−1 的气相加氢反应,条件为:温度160℃,n(H2)/n(F7−1)=10,接触时间22 s,连续运行500 h。结 果 显 示,500 h 内,F7−1 转 化 率 约 为99.5%,F7A 选择性约为99.0%[23]。该路线具有转化率高、选择性好、催化剂使用寿命长的特点。
F7−1 气相催化加氢反应机理是符合“先加成、后取代”机制[20],还是符合“先取代、后加成”机制[52],这样的争议一直存在。本课题组设计了以下方案,合成得到了顺式−1−氯−2,2,3,3,4,4,5−七氟环戊烷(cis−F7A−1Cl):在催化剂Pt+Bi+Au/AlF3存在下,温度150℃,n(H2)/n(F7−1)=5,接触时间20 s,连续运行1500 h,催化剂的活性比较平稳,1500 h 时,F7−1 转化率为49.3%,cis−F7A−1Cl 选择性为22.3%,F7A 选择性为47.9%,F6A 选择性为25.7%,F7E 选择性为3.1%。通过GC−MS 和核磁鉴定,证明了F7−1 加氢反应中确实存在含量比较大的加成产物cis−F7A−1Cl。cis−F7A−1Cl 的大量存在,证实了F7−1 的气相催化加氢机理符合“先加成、后取代”的机制[23,52]。
1.5 F6A的合成
F6A 的传统合成方法是采用F6−12 或1,5−二碘−2,2,3,3,4,4−六氟戊烷的液相氢解合成得到。Sekiya等[53]报道了以F6−12和氢气为原料,在高压容器中,三乙胺作为缚酸剂,在Pd/C 催化剂作用下合成F6A(沸点84.5~85℃),不过该方法涉及高压反应,对设备要求高,并使用溶剂,形成大量三废且后处理困难。Sekiya 等[54]以F6−12 和氢气为原料,在Pd/C 催化作用下,通过高压液相催化加氢反应合成F6A,该路线对设备要求严格,操作复杂,且不能满足连续生产。Sekiya等[55]以F6−12为起始原料,以石墨粉为载体制备的5%Pd/C 催化剂作用下,三乙胺作为缚酸剂,在高压釜中通入氢气,40℃下反应7 h,合成F6A。Leck 等[56]以F6−12 为起始原料,以镍为催化剂,同时以溶解有三乙胺的甲醇溶液为溶剂,在高压釜中通入氢气,在60~70℃下,反应制备F6A,收率60%。Michael 等[57]以1,5−二碘−2,2,3,3,4,4−六氟戊烷和三丁基氢化锡为原料,40℃下反应2 h,经后处理得到F6A,收率为79%。上述方法存在以下缺陷:(1)液相法制备F6A 的过程中需要加入缚酸剂或溶剂,后处理的过程中会产生大量的废液,该制备方法在实际的工业应用中所产生的大量废液造成环境污染,处理成本较高;(2)使用1,5−二碘−2,2,3,3,4,4−六氟戊烷为原料的制备方法,需要使用三丁基氢化锡,其毒性较大,过程不可控,并且产率较低,不适合工业化生产。
为解决上述难题,开发了气相反应制备F6A 的路线。在加氢催化剂1.0%Pd/AlF3存在下,F6−12与氢气发生气相催化加氢反应,条件为:温度200℃,n(F6−12)/n(H2)为1/7,接触时间18 s,连续运行10 h,F6−12 的转化率为100%,F6A 选择性为99.4%[52]。在上述反应中,F6−12 可以被替换为1−氯−3,3,4,4,5,5−六氟环戊烯(F6E−1Cl)或F6E,反应结果很类似,主要产物仍然是F6A。
1.6 cis-F8A的合成
传统合成cis−F8A 是采用直接氟化反应或者液相加氢反应合成得到。Burdon 等[58]以反式−1,2,3,3,4,5−六氟环戊烯(trans−F6E−34H)为起始原料,以三氟化钴为氟化试剂,200℃下进行反应,cis−F8A 收率为8.2%。该路线存在原料难以获取、产物收率过低的不足,不适合工业化生产。Takada 等[40]以1,2−二氯八氟环戊烷为起始原料,以LiAlH4为氢源,乙醚为溶剂,反应4 h 后,进行后处理,得到F8A,收率为75%。上述方法需要使用溶剂和氢化铝锂,比较苛刻的反应条件以及大量溶剂的使用导致成本昂贵,不利于工业化生产的实现。
为解决上述难题,开发了气相反应合成cis−F8A的路线。F8E 与氢气发生气相催化加氢反应得到cis−F8A。反应条件为:催化剂1.0%Pd/AlF3,反应温度200℃,n(F8E)/n(H2)为1/7,接触时间9 s,连续运行10 h,F8E 的 转 化 率 为100%,cis−F8A 选 择 性 为98.6%[52]。
1.7 F6E的合成
F6E可由F7A发生脱氟化氢反应得到。在DMF溶剂中,F7A 在160℃反应6 h,得到F6E 和F6E−15H,F7A 的 转 化 率 为83.3%,F6E 的 选 择 性 为97.0%,F6E−15H 的选择性为2.5%。在铬基催化剂存在下,反应温度300℃,F7A 的接触时间为60 s,反应压力为0.1 MPa,运行50 h,F7A 转化率为100%,F6E 选择性为93.2%,F6E−15H 选择性为6.8%[59]。在同分异构体F6E 和F6E−15H 中,F6E−15H 的化合物能量高于F6E[23],因此,F6E 属于比F6E−15H 更稳定的构型。这可以解释F7A 脱氟化氢反应中,F6E的选择性远大于F6−12H。在氟离子催化作用下,F6E−15H 向F6E 的异构化是不可逆的[60]。因此,可以通过进一步的氟离子催化,使F6−15H 彻底转化为F6E[28]。上述脱氟化氢路线可以高转化率、高选择性地合成F6E。然而在实际生产中,由于原料涉及到F6−12 气相氟化合成F7−1 属于氟化难度相当大的步骤,导致原料F7A 成本很高,因此,脱氟化氢路线存在生产成本过高的缺陷。
另外,F6E 可以通过F6−12 的一步气相加氢得到。在Cu/C催化剂存在下,n(F6−12)/n(H2)为1/1.09,反应温度260℃,接触时间125 s,则F6−12 的转化率为64.5%,F6E的选择性为20.6%,F6E−1Cl的选择性为76.1%[61]。当 在Ni/C 催 化 剂 存 在 下,n(F6−12)/n(H2)为1/8.64,温度360℃,接触时间22 s,运行6 h后,F6−12 的 转 化 率 为98.8%,F6E 的 选 择 性 为88.0%,F6E−1Cl的选择性为6.3%[61]。该路线存在反应温度过高、镍或铜催化剂在加氢脱氯反应中会快速失活,且多微孔结构的活性炭容易被积炭堵塞导致催化剂失活的缺陷[62]。
为解决上述难题,开发了F6−12 先选择性加氢或液相水解得到中间体F6E−1Cl、F6E−1Cl 再选择性加氢得到F6E 的工艺路线。首先,在加氢催化剂Pd/ZnF2存在下,反应温度200℃,n(F6−12)/n(H2)为1/10,接触时间15 s,连续运行200 h,F6−12 的转化率为85.6%,F6E−1Cl 选择性为83.3%,F6E 选择性为10.3%,F6A选择性为6.4%[63]。该气相选择性加氢路线不但反应温度较低,而且催化剂的使用寿命较长,容易实现气相连续法联产F6E−1Cl、F6E和F6A。
F6E−1Cl 也可以通过F6−12 的水解反应制备得到,即在搅拌条件下,在装有冷凝管、气球密封冷凝管出口的200 ml玻璃烧瓶中,加入锌粉、水、N,N−二甲基乙酰胺(DMF)和F6−12,n(Zn)/n(H2O)/n(DMF)/n(F6−12)为1.25/1/6.23/1,锌粉为0.25 mol,水为0.2 mol,DMF 为1.245 mol,F6−12 为0.2 mol,反应温度110℃,反应时间6 h。反应结束后,冷却至室温,用200 ml 水洗涤、过滤除去固体氢氧化锌和过剩的锌粉,将滤液进行常压蒸馏,得到F6E−1Cl(沸点为72~73℃/760 mmHg, 1 mmHg=133.322 Pa),收 率 为97.6%,纯度为99.7%[64]。其反应机理如下(图4):(1)DMF 结构中,与羰基相连的氢属于活泼氢,在加热条件下,锌粉与携带活泼氢的C—H 键发生插入反应,形成H−Zn−C(O)N(CH3)2中间体[65];(2)在DMF溶剂中,在中间体C−Zn−H 作用下,F6−12 容易释放出氯离子,形成不饱和的环状氯离子[66],同时H−Zn−C(O)N(CH3)2与氯离子反应生成Cl−Zn−C(O)N(CH3)2和氢负离子;(3)氢负离子进攻环状氯离子生成F6E−1Cl;(4)Cl−Zn−C(O)N(CH3)2与水发生水解得到DMF、氢氧化锌和氯化锌。因此,该反应实质上是DMF 作为催化剂催化F6E−1Cl 的水解反应。产物F6E−1Cl能否进行进一步的水解反应?答案是否定的。F6E−1Cl 如果要发生水解反应,前提是F6E−1Cl 离去氯离子,生成氢鎓离子,然而C—H 是非极性共价键,不存在孤对电子,导致氢离子[C−H−C]+是不存在的[66−67]。该路线对F6E−1Cl的选择性很高,不足之处是使用了大量溶剂和锌粉,容易产生液废和固废。
在F6−12 的气相选择性加氢和液相水解合成F6E−1Cl 的路线中,综合考虑转化率、选择性以及“三废”等因素,在工业上优选气相选择性加氢路线生产F6E−1Cl。
F6E−1Cl 经气相催化加氢反应合成F6E:在加氢催化剂Pd/ZnF2存在下,反应温度200℃,n(F6E−1Cl)/n(H2)为1/10,接触时间15 s,连续运行200 h,F6E−1Cl 的转化率为100%,F6E 选择性为92.4%,F6A 选择性为7.6%[63,68]。该路线采用F6−12 为起始原料,经历两步气相选择性加氢合成F6E,通过控制反应条件可以提高转化率、高选择性地合成F6E。而传统的一步气相加氢路线中,主要产物是F6A,F6E−1Cl 选择性不到1%,F6E 的选择性为零[52]。因此,F6−12 的两步气相选择性加氢是合成F6E 的较适宜路线。
1.8 F7E的合成
F7E可以通过F8E的加氢脱氟制备得到。Feast等[69]报道了15℃下,四氢呋喃为溶剂,八氟环戊烯与氢化铝锂发生加氢脱氟,反应4 h,生成F7E 以及3H−七氟环戊烯(F7E−3H)。
F7E 也可通过1−氯−1,2,2,3,3,4,4−七氟环戊烷(F7A1Cl−55H)脱氯化氢制备得到。Sugimoto[70]报道了以F7A1Cl−55H 为原料,催化剂为四(正丁基)溴化铵,溶剂为碳酸钾水溶液,50℃反应5 h,经过液相脱氯化氢反应合成F7E。Sugimoto 等[71]报道了采用F7A1Cl−55H 为原料,在加氢催化剂存在下,发生气相催化脱氯化氢反应,合成F7E。(1)原料F8E、cis/trans−F8A−12H 或F7A1Cl−55H 难以获取,且市场价格极为昂贵;(2)大多报道合成中间体F7E的工艺采用液相法,需要使用大量反应溶剂和大量难以回收的脱卤化氢试剂或脱卤试剂,产生大量废液,将严重污染环境。
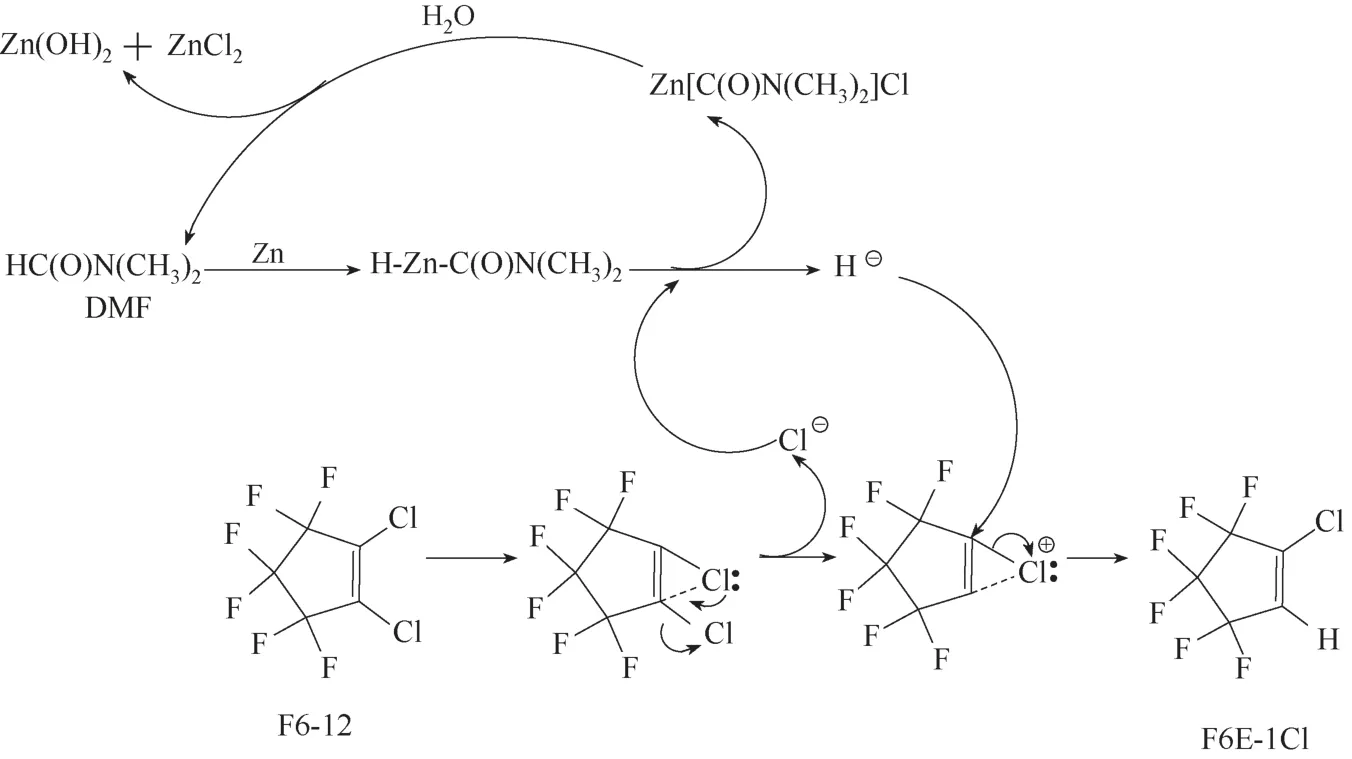
图4 DMF催化F6−12水解的反应机理Fig.4 Mechanism for DMF catalyzed hydrolysis of F6−12
F7E还可以通过顺式/反式−1,1,2,2,3,3,4,5−八氟环戊烷(cis/trans−F8A)脱氟化氢制备得到。通常情况下,采用液相反应工艺。Heizman 等[72]报道了cis/trans−F8A 在氢氧化钠水溶液中回流1 h,得到产物F7E 和F7E−3H,其沸点依次为46℃和51℃。Banks等[73]报道了室温条件下,cis−F8A 在四氯乙烯溶剂中,碱性离子交换树脂存在下反应1.5 h,转化率为60%,F7E 的选择性为83.3%,F7E−3H 的选择性为16.7%。Sugimoto[74]报道了在碳酸钾水溶液中,以cis/trans−F8A 为原料,四(正丁基)溴化铵为催化剂,45℃反应7 h,转化率为97.9%,F7E、F7E−3H 和4H−七氟环戊烯(F7E−4H)的选择性依次为89.9%、9.5%、0.6%。在三种同分异构体中,按照化合物能量由大到小排序为:F7E−3H >F7E−4H >F7E,三者中F7E属于最稳定构型[23]。因此,在cis−F8A 或cis/trans−F8A 脱氟化氢反应中,F7E 的选择性远大于其他两者。在氟离子催化作用下,F7E−4H 与F7E−3H 之间的异构化是可逆的,而F7E−3H 向F7E 的异构化则是不可逆的[60]。研究认为,在DMF 溶剂中,氟离子催化F7E 或F6E 的同分异构体发生异构化反应,异构体的相互转化主要包含两种途径:一种是烯丙基对氟离子的顺式−加成/消去机制,该机制并不能改变氢原子的相互位置,但是负责氟离子的迁移;另一种是氟离子辅助的去质子化/质子化机制,该机制并不能改变氟原子的相互位置,但是负责氢原子的迁移(图5)[60]。同时,还揭示了来自于去质子化过程的HF2−阴离子可以抑制质子化过程。这可能是平衡常数的实验值总是和来自于量子化学计算的理论值不一致的原因。由于液相脱氟化氢制备F7E存在三废多、工艺间歇难以实现连续性等缺点,因此不属于理想的制备方法。
F7E 可以通过F6−12 气相氟化得到F7−1、F7−1气相选择性加氢得到。其中,气相氟化步骤的最高收率为18.0%[22],气相选择性加氢步骤的最高收率为67.3%[75],则F7E 的单程最高总产率仅为18.0%,特别是在第二步反应中,F7E的选择性很低,有大量的副产物F7A、F6E和F6A生成[75]。
为解决上述难题,开发了cis−F8A 气相脱氟化氢制备F7E 的工艺路线。在铬基催化剂存在下,反应温度300℃,cis−F8A 的接触时间为60 s,反应压力为0.1 MPa,运 行50 h,cis−F8A 转 化 率 为98.1%,F7E−4H 选择性为0.1%,F7E 选择性为99.7%,F7E−3H 选择性为0.2%[59]。在三种同分异构体中,按照化合物能量由大到小排序为:F7E−3H >F7E−4H >F7E,三者中F7E 属于最稳定构型[23]。因此,在cis−F8A 脱氟化氢反应中,F7E 的选择性远大于其他两者。可以通过异构化催化剂催化F7E−3H 和F7E−4H 彻底转化为F7E[28]。该路线存在cis−F8A 原料成本过高的缺陷。

图5 氟离子催化的氢氟环戊烯异构化反应的两种途径Fig.5 Two ways of hydrofluorcyclopentene isomerization catalyzed by fluoride ion
另外,开发了F6E−1Cl 气相催化氟化反应合成F7E 的工艺路线。在铬基催化剂存在下,反应温度420℃,n(F6E−1Cl)/n(HF)为1/20,接触时间8 s,连续运行20 h,F6E−1Cl 的转化率为62.7%,F7E 选择性为97.2%[63,76]。该路线采用F6−12 为原料经气相选择性加氢、气相氟化的两步路线合成F7E,F7E 的单程总产率较高(43.5%),特别是在第二步反应中,F7E的选择性很高,在后续工序中容易分离,不会造成原料的浪费。因此,F6−12经气相选择性加氢、气相氟化共两步反应是目前合成F7E的较适宜路线。
2 五元环氟化物的应用
2.1 电子清洗剂
电子清洗领域,是关乎国家重大战略的行业,涉及到半导体、电子、航海、航空、太空等行业的精密仪器的清洗。依据其环保参数、产品特点和发展历程,目前把高端清洗剂一般划分为四代。第一代清洗剂是对臭氧层具有破坏作用的CFCs 或氢氯烃(HCCs)类清洗剂,包括1,1,2−三氟−1,2,2−三氯乙烷(CFC−113)、四氯化碳(CTC)、三氯乙烷(TCA)。第二代清洗剂是HCFCs,包括1,1−二氯−1−氟乙烷(HCFC−141b)、1,3−二 氯−1,1,2,2,3−五 氟 乙 烷(HCFC−225ca/cb)。第三代清洗剂是HFCs、氢氟醚(HFEs)或碳氢类清洗剂,包括1,1,1,3,3−五氟丁烷(HFC−365mfc)、1,1,1,2,2,3,4,5,5,5−十氟戊烷(HFC−4310mee)、1,1,1,2,2,3,3,4,4−九氟−4−甲氧基丁烷(HFE−449s1)。作为CFCs 替代物的HFCs 类化合物被公认为具有最优良的清洗性能,虽然这类物质不破坏大气臭氧层,但大部分物质具有较高的GWP值[6−9]。因此寻找具有低GWP值,且满足优异清洗性能,适用的HFCs类清洗剂成为当务之急。
研究认为,F7A、F6A、cis−F8A 为代表的五元环氟化物可作为HFCs 类清洗剂的替代物[10−11]。其中,F7A 的ODP 值为0、GWP100值较低(195)、低毒、不燃,环境友好,清洗性能优良,被认为是有发展潜力的新一代清洗剂中的代表产品之一,可广泛应用于电子、邮电、航空航天、轻工、纺织、机械、医疗、汽车、精密仪器等行业,尤其在电子清洗中占有重要地位。F7A 不仅可应用于清洗领域来清洗光敏燃料[77]和作为溶剂在制备聚合物膜燃料电池中应用于能源领域[78],F7A 最重要的用途是作为清洗剂应用于清洗行业[79−80]。
HCFC−141b、 HFC−365mfc、 HFC−4310mee、HFE−449s1 是在国内外市场上占据主导地位的清洗剂,这些清洗剂与F7A 的性能比较见表1。由表1可知,F7A 的基本物理性能与HCFC−141b 相似。F7A 的沸点为82.5℃,在此温度下清洗零件,F7A 不会对温度敏感部件或材料产生影响;F7A 的贝壳松脂丁醇值(KB 值)=14(混合工质的KB 值可达20),在不含氯的含氟溶剂中,该值是最高的,说明F7A溶剂的溶解能力均衡,既可除油、油脂和脏污,又不会损坏金属或弹性零件[81]。F7A 的表面张力较小,浸透性好,可以浸入毛细部及缝隙间,升高温度能提升对油的溶解性,蒸汽的清洗效果非常好。汽化潜热是清洗剂是否能用于气相清洗的一个主要判断依据。F7A 的汽化潜热比较低,使溶剂汽化的耗能较少,适用于气相清洗。另外,F7A的环境性能要大大优于HCFC−141b[23]。而F7A 与HFC−365mfc相比,两者的KB 值相当,但F7A 的大气寿命要远低于HFC−365mfc,且F7A 的不可燃特性使其在使用过程中安全性大幅度提升。F7A 与HFC−4310mee相 比,F7A 的GWP100值 为195,远 低 于HFC−4310mee。F7A与HFE−449s1相比,F7A的洗净能力要强于HFE−449s1,故F7A 具有较强的商业竞争力[23]。DeGroot 等[82]开发了一种新型共沸清洗剂的配方,其中复配物包括1−溴丙烷、反式−1,2−二氯乙烯、二氯甲烷,乙醇、异丙醇等与氢氟环戊烷形成二元混合体系,可以低温清洗和漂洗相结合的方式。由于商业利益的原因,上述公开文献并未对F7A 的清洗配方和清洗性能进行系统的报道。在五元环氟化物中,F6A、F7A 和cis−F8A 分别与5%叔戊醇混配,分别对油污和电路板进行清洗。结果显示,F7A的清洗性能优于F6A和cis−F8A[83]。
2.2 刻蚀气体
芯片制造的核心技术之一在于对硅基材料的高精度刻蚀,因此对刻蚀气体不仅要求其形成的等离子体提供丰富的刻蚀粒子,同时还需提供形成阻挡层的前驱物分子以提高刻蚀的选择比和各向异性。目前,能满足使用要求的主要是碳氟化合物,比如四氟甲烷、六氟乙烷、八氟环丁烷等饱和全氟烷烃,然而饱和全氟烷烃的GWP100值很高,四氟甲烷、六氟乙烷、八氟环丁烷的GWP100值依次是6630、11000和9540,属于需要淘汰的高温室气体。因此,亟需设计和开发环境友好、刻蚀性能优异的刻蚀气体来替代全氟烃刻蚀剂。与四氟甲烷、六氟乙烷、八氟环丁烷相比,F8E 的GWP100值最低,仅为90。另外,F8E 对SiO2的刻蚀速率和刻蚀选择比优于八氟环丁烷[84](表2),被认为是替代上述饱和全氟烃的理想刻蚀剂之一。
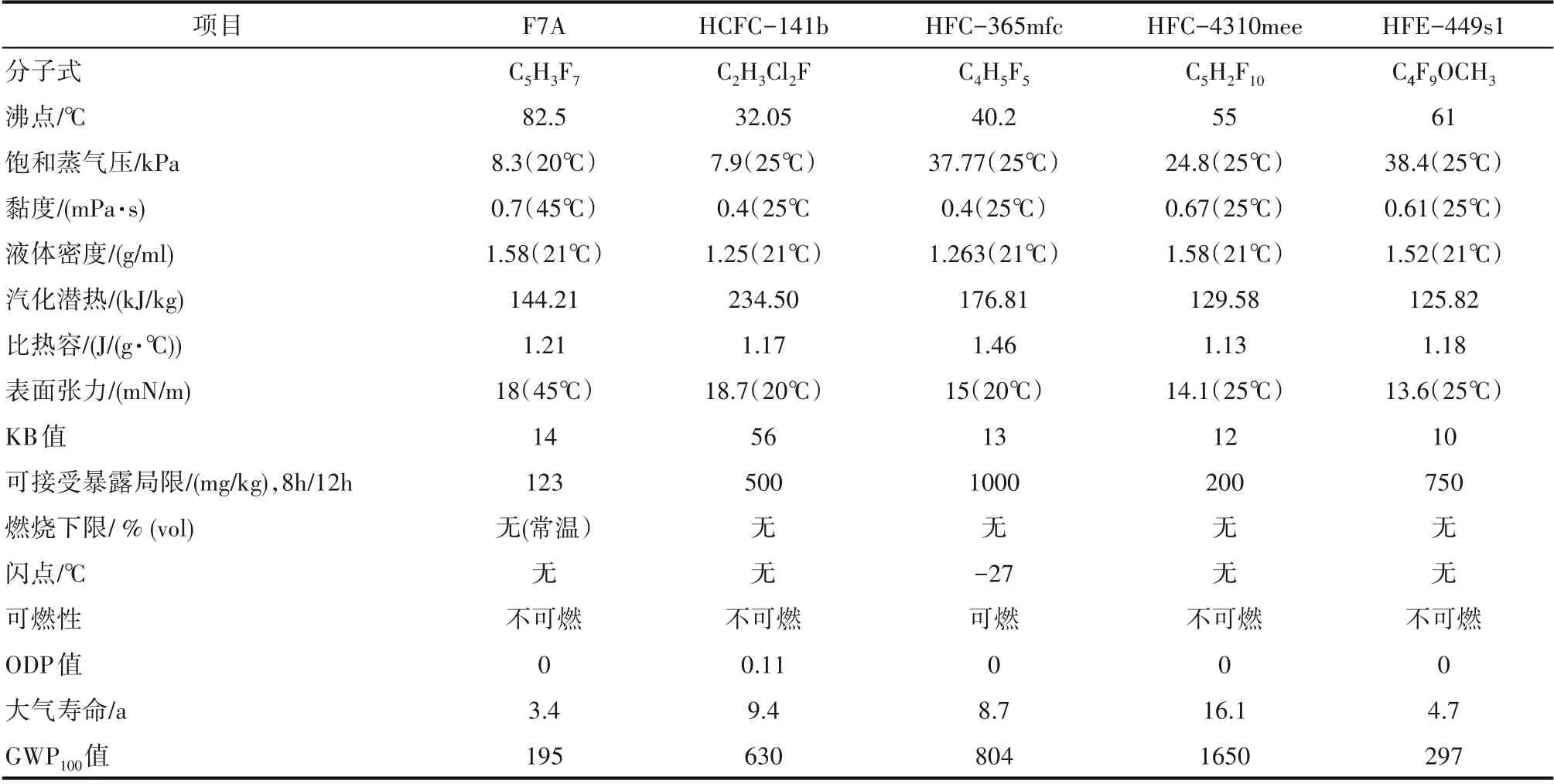
表1 各种清洗剂的基本性能[23]Table 1 Basic properties of various cleaning agents[23]
采用傅里叶变换质谱研究F8E 在10~7 Torr(1 Torr=133.322 Pa)压力范围内的单次碰撞条件下的离子化学性质。研究结果表明,低能(<40 eV)的离子群主要是C3F3+、C4F6+、C5F7+、C4F5+和C5F8+占优,Ar+与F8E 发生的电荷转移反应会产生重要的含氟物种,从而可用作刻蚀气体[13]。F8E 可以与氩气和氧气混合作为刻蚀气体,在Ar 1000 sccm/F8E 15 sccm/O228 sccm、2 Pa 等离子体、偏置功率400 W 的条件下,该刻蚀气体对二氧化硅的刻蚀速率为364 nm/min,对SiCN 的刻蚀速率为26 nm/min,二氧化硅对SiCN 的选择比为14,远高于CF4(CF4200 sccm、1 Pa等离子体、晶圆偏置100 W)作为刻蚀气体的选择比(4.9)[85]。研究认为,当F8E 分流率从38%增加到62%时,二氧化硅的蚀刻速率会增加,然后在F8E百分比高于62%时,二氧化硅的蚀刻速率突然下降。在高F8E 流速、低Ar 流速、高工艺压力和低RF 功率的条件下,Si3N4对二氧化硅的选择性会增加,氟碳聚合物的选择性也会增加[86]。添加CH2F2导致氮化物蚀刻速率的降低与F 自由基以及O 和CO 自由基的降低有关。在自对准接触孔(SAC)图案化晶圆中,向晶圆中添加了最适量的CH2F2/F8E/O2/Ar 产生明确的轮廓,可以明显改善厚沉积膜的台阶覆盖率,并且封端氮化物没有额外的损失[87]。
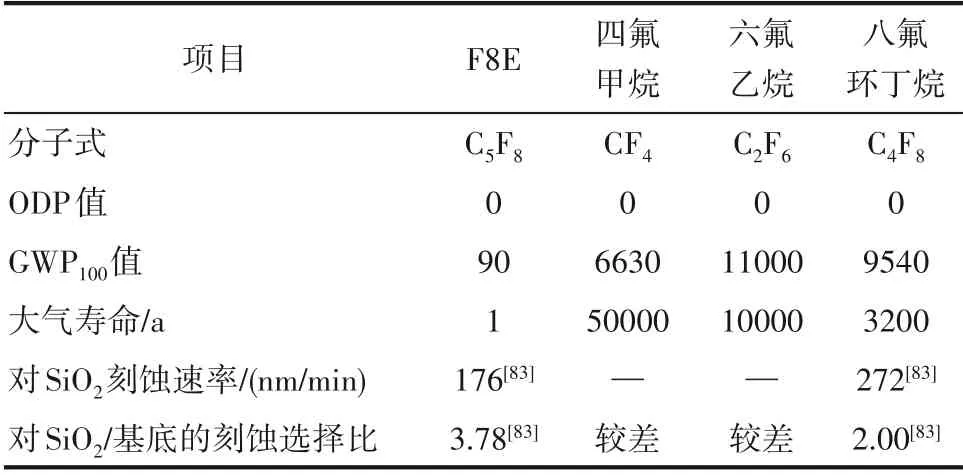
表2 各种刻蚀剂的基本性能Table 2 Basic properties of various etching agents
2.3 用于合成电子氟化液
2018 年,我国数据中心总用电量1608.89×108kW·h,远超三峡大坝1016×108kW·h 的年发电量,其中,数据中心的散热占电力消耗的比重巨大,采用空调系统给数据中心降温的能耗占用了整个数据中心能耗的1/3 以上。随着数据中心的规模越来越大,部署的机架越来越密集,对服务器负载的要求越加苛刻,传统的空气冷却技术由于散热效率低下而不再满足数据中心的制冷要求,往往会发生重大故障,轻则装置和设备烧毁,重则数据和信息尽失。
目前,浸没式液冷技术成了解决上述难题的有效方案。美国3M 公司、比利时索尔维公司等国外氟化工巨头分别推出了高介电常数、导热性好的全氟烷基胺、全氟聚醚等电子氟化液作为浸没式冷却液,用于数据中心的散热。上述电子氟化液存在以下缺陷:(1)现有电子氟化液的环境性能很差,全氟烷基胺和全氟聚醚的GWP100值一般大于5000,甚至更高,大气寿命长达数百年;(2)全氟烷基胺主要采用电解氟化的合成工艺,该方法存在合成困难、合成效率低的缺陷;(3)对于快速发展的数据中心规模来说,全氟烷基胺和全氟聚醚的传热效率比较低,冷却液使用量大。因此,全氟烷基胺和全氟聚醚都不是理想的电子氟化液。截至目前,市场上还没有一种电子氟化液同时满足高介电常数、低毒性、高导热性、高沸点、低倾点、低GWP100值、易合成的使用要求。
研究认为,环骨架的含氟醚和不饱和全氟胺可作为电子氟化液全氟烷基胺的潜在替代物[88−90]。环骨架的含氟醚和不饱和全氟胺可以通过卤代环烯烃(卤=氟或氯)与含氟醇或全氟亚胺反应得到。
在非质子溶剂中,碱作用下,卤代环烯烃(卤=氟或氯)为原料与含氟醇先发生亲核取代反应,再经过系列反应得到环骨架的含氟醚,包括式(Ⅰ)和式(Ⅱ)的结构(图6)。在合成过程中,通过控制反应原料的物料比,容易依次实现烷氧基在卤代环烯烃上对1 位、2 位、3 位碳位上的卤原子进行取代,获得具有不同取代基数量的含氟醚,调控其沸点;其次,可通过控制含氟醇的链长,来调控含氟醚的沸点;再其次,可通过控制醇的氟原子或氢原子数量,来调控含氟醚的可燃性、介电常数、大气化学性质等物化性质。与现在使用的全氟烷基胺(GWP100值>5000)相比,环骨架的含氟醚具有高介电强度、低毒性、优异的传热性能、高沸点,且GWP100值仅为全氟烷基胺的数十分之一的数量级甚至更低[88]。因此,环骨架的含氟醚是性能优良的电子氟化液。
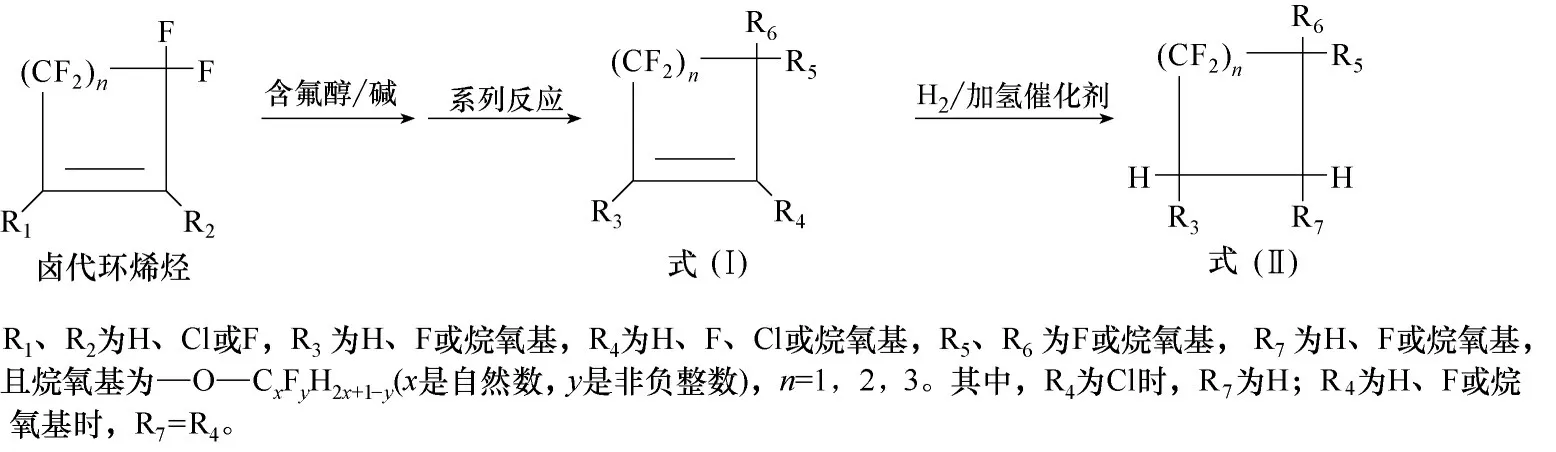
图6 环骨架的含氟醚的合成路线Fig.6 Synthetic route of fluorinated ether with ring skeleton
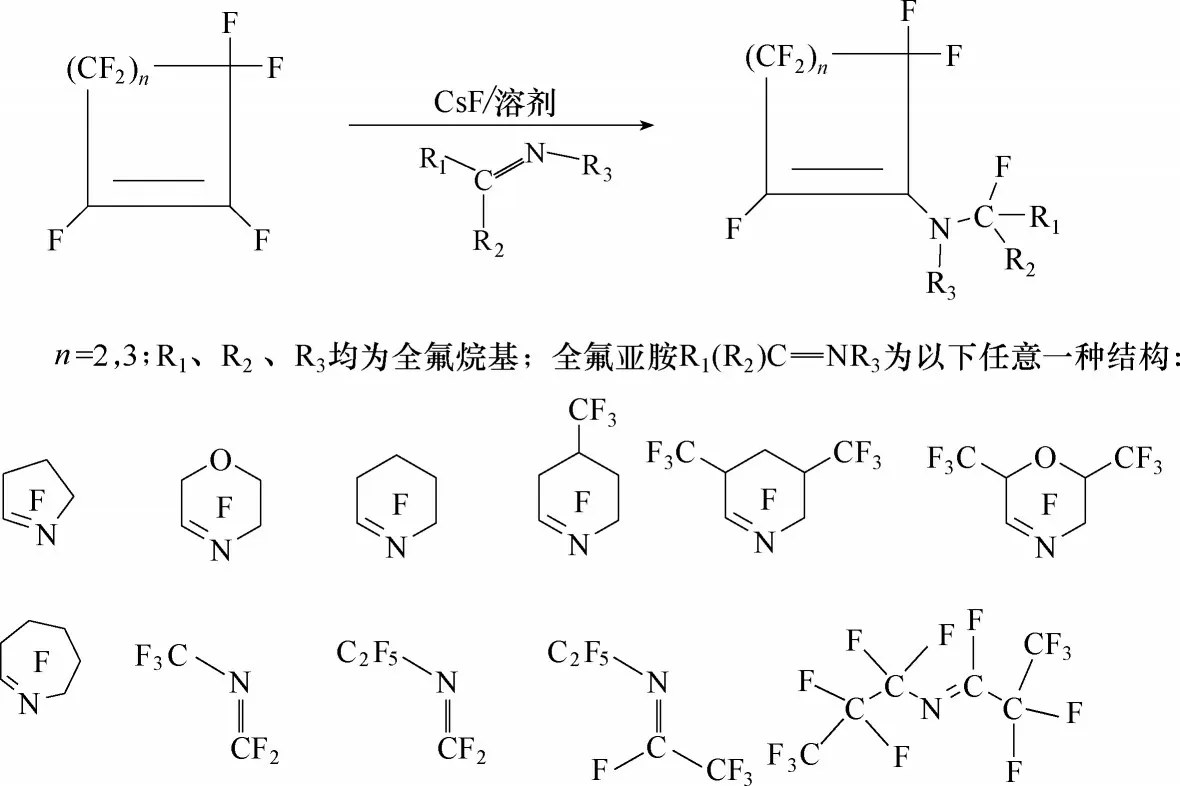
图7 环骨架的不饱和全氟胺的合成路线Fig.7 Synthetic route of fluorinated amine with ring skeleton
在二甘醇二甲醚溶剂中,氟化铯催化作用下,全氟环烯烃与全氟亚胺发生亲核取代反应,得到环骨架的不饱和全氟胺(图7)。在合成过程中,通过调控原料全氟亚胺的类型以及全氟环烯烃的碳原子数量,来调控目标产物的倾点以及其他特性。比如:2,2,3,3,4,4,5,5−八氟−1−(全氟环己烯基)吡咯烷(PE−1)、2,2,3,3,4,4,5,5−八氟−1−(全氟环戊烯基)吡咯烷(PE−2)、2,2,3,3,5,5,6,6−八氟−4−(全氟环戊烯基)吗啉(PE−3)三种替代物的倾点分别为−34℃、−33℃和−49℃。其中,PE−1 的GWP100值=416,远小于一般饱和全氟烃的GWP100值(大于5000),介电常数是1.98,与目前正在使用的三(全氟正丁基)胺(FC−43)和三(全氟正戊基)胺(FC−70)的介电常数相当(表3)[90]。因此,环骨架的不饱和含氟胺被认为是替代目前电子氟化液全氟烷基胺的潜在工质之一。
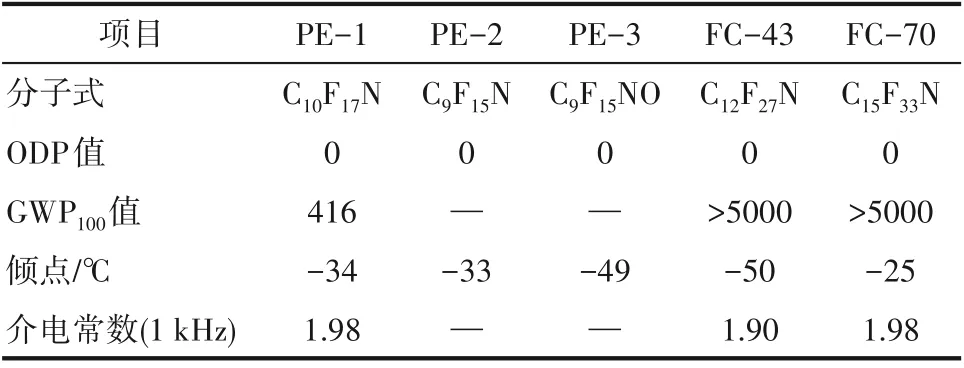
表3 环骨架的不饱和含氟胺与全氟烷基胺的基本性能Table 3 Basic properties of unsaturated fluoroamines with ring skeletons,and perfluoroalkylamine
3 结 论
在五元环氟化物的合成研究中,综合考虑原料易得且廉价、高效合成、对环境近零污染的原则,目前较佳的合成路线如下:以DCPD 为起始原料,经气相热解、CPD 的液相氯化、TCCPA 的气相催化氯氟化得到F6−12;F6−12 的气相催化氟化得到F7−1;F7−1的气相催化氟化得到F8E;F7−1的气相催化加氢脱氯得到F7A;F6−12 的气相催化加氢脱氯得到F6A;F8E 的气相催化加氢得到cis−F8A;F6−12 的气相催化选择性加氢得到F6E−1Cl,F6E−1Cl 的气相选择性加氢得到F6E;F6E−1Cl 的气相催化氟化得到F7E。上述路线和技术已经被国内公司采用,建成了世界首套工业化联产五元环氟化物的连续生产线。目前,五元环氟化物的合成研究重点是新型合成路线的开发,高催化活性且对人体无害的过程催化剂的开发,以及无污染化工艺过程的开发。其中,在氟化工中,铬基催化剂由于原料易得、催化活性较高、使用寿命长的优点,成为氟化工过程中最重要的过程催化剂,但存在难以回收再利用的缺陷,而且在一定条件下三价铬离子可以转化为高致癌性的六价铬离子。六价铬被世界卫生组织国际癌症研究署(IARC)列为“人类致癌物”。因此,开发出对人体健康无害、高催化活性、长使用寿命的非铬催化剂来替代目前仍在广泛应用的铬基催化剂显得尤为必要。
在五元环氟化物的应用研究中,目前的研究重点是五元环氟化物电子级产品的开发,以及设计与开发新型的五元环氟化物的下游产品。其中,F7A、F8E分别用作电子领域的清洗剂,其纯度越高越好。目前市场上F7A 的纯度可达到99.9%,F8E 的纯度可以达到99.99%,成品里面往往还有10−6级的含氯化合物杂质,这类含氯化合物在清洗或刻蚀过程中往往会毁坏电子原件或设备。因此,开发出更高纯度的电子级产品显得尤为重要。另外,通过发展五元环氟化物的下游产品,找寻具有特殊性能的新型功能含氟材料,从而在国际先进氟材料领域中占有一席之地。
