全自动免疫亲和固相萃取超高效液相色谱法测定粮油中黄曲霉毒素
2020-09-03王海波轩志宏刘洪美王松雪何建洪
李 丽 吴 宇 王海波 叶 金 轩志宏刘洪美 王松雪 张 峰 何建洪
(国家粮食和物资储备局科学研究院1,北京 100037) (广西-东盟食品检验检测中心2, 南宁 530021) (睿科仪器(厦门)有限公司3,厦门 361006)
黄曲霉毒素(Aflatoxins,AFT) 是黄曲霉、寄生曲霉等代谢产生的一类结构相似的化合物,常见的有黄曲霉毒素B1、B2、G1、G2四种,其中以黄曲霉毒素B1的毒性最大,被世界卫生组织列为I类致癌物。黄曲霉毒素广泛存在于玉米、稻谷、花生、棉籽及一些坚果类食品和饲料中。对人畜有强烈的致病性、致癌性,是危害最严重的真菌毒素之一,受到世界各国的广泛关注[1-3]。鉴于黄曲霉毒素对人类的的危害,我国在2016年颁布的《食品中真菌毒素限量》分别规定了粮油中黄曲霉素B1的限量值[4]。
黄曲霉毒素的检测方法有很多种,其中薄层色谱法[5]由于溶剂消耗量大、重现性差等原因使用较少,酶联免疫吸附法[6]和胶体金试纸条法[7]多用于黄曲霉毒素的快速筛查,液相色谱质谱联用法[8-11]主要用于多种真菌毒素同时监测,但由于仪器昂贵,使用成本较高。免疫亲和柱具有有机溶剂消耗少、基质干扰少、特异性强、灵敏度高和适用于各种复杂基质的样品等特点[12,13],因此免疫亲和层析净化高效液相色谱法是目前国际上普遍采用的黄曲霉毒素精确定量和确认的方法[14-16]。大部分实验室对样品中黄曲霉毒素检测前处理时采用手动净化。而手动净化由于过程繁琐、耗时长等因素制约,导致每天处理样本量少。当样品量较大时,存在时效性差、人为因素影响大等问题。S. Jinap等[17,18]建立了免疫亲和柱在线净化高效液相色谱法检测花生、无花果干、红辣椒粉等样品中的黄曲霉毒素,但该方法干扰较多,样本量大时,容易堵塞色谱柱,且处理结果时需要扣除空白才能定量,处理数据方法较为复杂。
基于上述问题,本研究采用全自动固相萃取技术,结合免疫亲和柱建立了全自动免疫亲和柱固相萃取超高效液相色谱法高通量检测粮油中黄曲霉毒素的方法。本方法简化了前处理过程,提高检测的自动化水平和检测效率,可以满足大规模样品准确定量的需求。
1 材料与方法
1.1 仪器与试剂
Fotector Plus全自动固相萃取仪,Waters I-Class超高效液相色谱仪,AutoEVA-60全自动浓缩氮吹仪,固相萃取仪同,Evergreen TREA黄曲霉毒素免疫亲和柱。
甲醇、乙腈:HPLC级;PBS盐包、曲拉通-100:分析纯;国家有证标准物质GBW(E)100302甲醇中黄曲霉毒素B1;黄曲霉毒素B2、G1、G2标准品(纯度≥99%);自然污染黄曲霉毒素有证标准物质和质控样品:花生油中黄曲霉毒素B1质控样、大米粉中黄曲霉毒素B1和玉米赤霉烯酮质控样、玉米粉中黄曲霉毒素B1标准物质GBW(E)100386和玉米全粉空白(DON、ZEN、AFS)。
1.2 实验方法
1.2.1 样品准备
配制高、中、低三种浓度黄曲霉毒素玉米、糙米和花生油加标样品。
自然污染真菌毒素有证标准物质和质控样品直接称量使用。
1.2.2 仪器条件
WATERS I CLASS 配荧光检测器(大体积流通池)。
色谱柱:Waters BEH-C18柱(2.1 mm×100 mm,1.7 μm),柱温:40 ℃。
流动相:A相:乙腈∶甲醇(体积比为1∶1);B相:水,A∶B=35∶65,等梯度洗脱。
流速为0.3 mL/min,进样量为10 μL。
激发波长:365 nm;发射波长463 nm。
1.2.3 样品前处理方法
准确称取(5±0.01) g样品,加入20 mL 84/16乙腈/水,振荡提取30 min或均质3 min,7 000 r/min离心5 min或定性滤纸过滤,取4 mL上清液加入46 mL 0.1% PBST缓冲液,取全部稀释液于全自动固相萃取仪80 mL上样架上,运行编辑完成的仪器方法对样品进行自动净化(具体步骤详见表1)。将洗脱液直接与收集架一起转移到全自动氮吹仪内于50 ℃以下氮气吹干,用1 mL流动相溶解残渣,供液相色谱测定。

表1 全自动固相萃取步骤
2 结果与讨论
2.1 免疫亲和柱的性能评价
免疫亲和柱是粮油食品检测净化的关键耗材。但是,由于免疫亲和柱生产工艺不同,导致各品牌免疫亲和柱产品性能和质量差异较大。因此,需要对免疫亲和柱性能进行评测,主要考察免疫亲和柱的柱本底、柱容量和柱回收等参数。
本研究选取的亲和柱柱本底未检出、柱容量满足标准GB 5009.22—2016《食品中黄曲霉毒素B族和G族的测定》相关要求的3个品牌亲和柱开展柱回收评价实验。
基于实际检测的要求,考察定量限附近、限量值、标准曲线上限附近三个浓度水平,按照3水平6平行的原则,每批次随机抽取18根进行评测。按照1.2.3的方法处理玉米粉阴性样本,获得玉米粉阴性样本的稀释液,在稀释液中添加一定量的黄曲霉毒素综合评测免疫亲和柱的精确性。结果如表2所示。

表2 不同品牌黄曲霉毒素免疫亲和柱柱回收评价结果
从表2可知,品牌A和品牌C黄曲霉毒素免疫亲和柱对AFB1、AFB2、AFG1、AFG2四种黄曲霉毒素的回收率均在85%以上,相对标准偏差小于4.6%,满足实验需要。但是B品牌的黄曲霉毒素免疫亲和柱对AFG2的吸附率较低,回收率低于50%,不适用于本方法的研究。在本实验中我们随机选取同一批次A品牌的免疫亲和柱开展后续研究。
2.2 稀释液的优化
由于免疫亲和柱对有机溶剂的耐受性是有限的,所以样品经提取液提取后,需对提取液进行稀释才可以过柱。考虑到粮食样本中玉米基质较为复杂,所以分别用水、磷酸盐缓冲液(PBS)、0.1 %曲拉通-磷酸盐缓冲液(0.1 %PBST)三种稀释液稀释玉米粉提取液,考察稀释液对检测结果准确性的影响,结果见表3。

表3 不同稀释液的比较(n=6)
通过t检验,两两之间没有显著区别,但在实际操作过程中,由于粮食基质较为复杂,提取后用水或者PBS稀释,稀释液比较浑浊,过滤后也没有明显的改善。但是加入0.1% PBST后,稀释液变得澄清,不容易堵柱,谱图杂峰干扰少。所以选择0.1%PBST作为稀释液。

图1 上样流速对黄曲霉毒素回收率的影响(n=6)
2.3 上样流速的优化
为了缩短上样时间,同时保证待测毒素全部吸附到免疫亲和柱上,考察玉米空白基质加标稀释液在上样流速分别为每分钟2、3、5、7 mL时的柱回收率,结果如图1所示,流速为2~7 mL/min时,四种黄曲霉毒素的回收率均在95 %以上,均符合标准GB/T 27404—2008《实验室质量控制规范 食品理化检测》要求[19]。但是7 mL/min时,检测结果相对标准偏差较其他上样流速时较大,因此综合考虑,选择5 mL/min的上样流速,确保时效性、回收率及精密度最好。
2.4 与手动法检测结果的对比
采用本方法和手动法分别对配制的高、中、低三个浓度梯度的玉米加标样品进行双实验检测,结果见图2,手动法和高通量全自动固相萃取法回收率均在78%~105%,RSD值小于7.0%,符合标准GB/T 27404—2008《实验室质量控制规范 食品理化检测》要求。采用配对t检验法比较两种方法的检测结果是否存在显著性差异,td小于临界值,说明两者之间无显著性差异。

图2 本方法和国标方法在玉米样品中加标回收率对比
分别采用本方法和手动法每3 h测定中浓度加标样品一次,连续测定18次。比较两种方法的重现重复性,结果见表4。本方法的重复性测定标准差和18次测定极差均优于手动法测定结果,稳定性良好。此外,与常规手动净化每人1 d处理20个样品相比,本方法1 d可净化并检测40个样品,显著提高了检测效率。

表4 本方法和国标法连续测定18次的加标回收率
2.5 方法的精密度与准确度考察
添加量为20 ng/g(以黄曲霉毒素B1计,其余3种毒素浓度按B1∶B2∶G1∶G2为4∶1∶4∶1混配)的同一批玉米样品重复测7 d,每天测6次,做方法精密度实验,结果见表5。采用本方法对添加黄曲霉毒素的玉米、糙米和花生油样品进行方法回收率检测。每个浓度做6 个平行样检测,结果见表6。本方法的日内精密度为1.8 %~3.0 %,日间精密度为1.8 %~4.7 %,加标回收率为75%~104%。

表5 精密度实验结果

表6 回收率实验结果
2.6 台间差考察
随机选择两台设备分别对花生油中黄曲霉毒素B1质控样(Peanut oiL-AFB1-2018)、大米粉中黄曲霉毒素B1和玉米赤霉烯酮质控样(RICE-ZEN&AFB1-2018)、玉米粉中黄曲霉毒素B1标准物质(GBW(E)100386)进行双实验检测,具体操作:质控样品经振荡提取后,提取液分成2份,分别用全自动固相萃取仪1和2配合免疫亲和柱净化处理,上机检测,结果见表7。

表7 台间差及方法准确度结果(n=6)
采用配对t检验法比较两台设备的检测结果是否存在显著性差异,两台仪器测定玉米粉中黄曲霉毒素B1(GBW(E)100386)结果统计td值为-0.347 8,小于临界值t0.01,5为4.032 1说明两台仪器间没有台间差检测结果无显著性差异。同理,大米粉中AFB1和花生油中AFB1质控样测定结果也可以得到相同的结论,满足LS/T 6402—2017《粮油检验 设备和方法标准适用性验证及结果评价一般原则》[20]的要求。同时,3个质控样的测定结果均在给定值不确定度范围之内,说明本方法检测结果准确可靠。
2.7 仪器空白考察
随着全自动固相萃取仪器使用次数的增加,有可能会有待测物残留的情况。因此每次处理样品前后对仪器空白的监测非常必要。图3是连续5次的仪器空白与标准品的对比色谱图,可以看出仪器无残留现象。

图3 仪器空白与标准品的对比图
2.8 方法不确定度考察
依据《化学分析中的不确定度的评估指南》[21]、 国家计量技术规范《测量不确定度评定与表示》[22],对本研究建立的全自动免疫亲和固相萃取超高效液相色谱法测定样品中黄曲霉毒素 B1含量进行不确定度分析评定,提高实验精度,为正确评价数据提供参考。
检测方法的不确定度分量主要有称量、移液等引入的不确定度、标准溶液引入的不确定度、回收率引入的不确定度和仪器稳定性引入的不确定度等(见图4)。

图4 不确定分量来源图
2.8.1 数学模型
根据已建立的检测方法,得出样品中黄曲霉毒素B1的含量计算公式。
式中:X为试样中待测毒素的含量/μg/kg;C为试样中待测毒素按照外标法在标准曲线中对应的浓度/ng/mL;V1为试样提取液的体积/mL;V2为用于净化移取的样品提取液/mL;V3为洗脱后最终定容体积/mL;m为试样称样量/g。
2.8.2 各分量不确定度的评定
2.8.2.1 B类不确定度uB
实验过程中,有些不确定度分量是由非统计学方法获得的,称之为B类不确定度。B类不确定度主要包括质量、体积等。在本研究中B类不确定度见表8。

表8 称量、移液等引入的不确定度分量
2.8.2.2 实验过程中黄曲霉毒素B1标准曲线引入的不确定度uc
黄曲霉毒素B1标准曲线引入的不确定度主要有标准溶液自身的不确定度、标准系列工作液配置和标准曲线拟合产生的不确定度3部分组成。
1)黄曲霉毒素B1标准溶液引入的不确定度uc-1
黄曲霉毒素B1溶液标准物质,GBW(E)100302浓度m1=1.96 μg/mL,扩展不确定为0.09 μg/mL(k=2),其相对标准不确定度是:
2)标准系列工作液配制引入的不确定度uc-2
黄曲霉毒素B1的标准系列工作液浓度为2、5、10、20、50 ng/mL,流动相定容。配制过程及引入的不确定度如表9所示。
根据表9可知,高效液相色谱法配制标准系列工作液时产生的相对不确定度为:
=1.74%
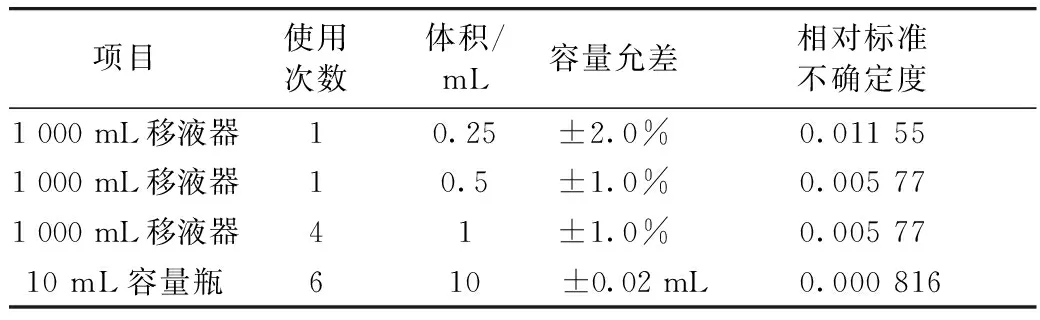
表9 标准系列工作液配制引入的不确定度
3)超高效液相色谱法中黄曲霉毒素B1线性拟合引入的不确定度uc-3
采用5个浓度梯度的黄曲霉毒素B1标准溶液,分别用UPLC重复测定3次,得到相应的峰面积,利用数据处理软甲拟合得到线性回归方程为Yi=a×Xi+b,式中:a为斜率;b为截距。测定结果及数据计算见表10。

表10 标准曲线数据
粮食样品中黄曲霉毒素B1重复测定6次,其测定结果见表11。

表11 黄曲霉毒素B1平行测定结果/ng/mL
则由工作曲线拟合所产生的不确定度计算公式如下:
式中:S为标准溶液峰面积残差的标准差,计算方法为:


2.8.2.3 方法回收率引入的不确定ure

表12 黄曲霉毒素B1加标回收测定结果
因此,回收率相对标准不确定度为:
2.8.2.4 黄曲霉毒素B1重复测量引入的不确定度urep
样品中黄曲霉毒素B1含量独立重复测定6次,测定结果见表11。
3.8.2.5 高效液相色谱仪器稳定性引入的不确定度us
2.8.3 标准不确定度的合成与扩展uX
将各分量合成,计算花生油中黄曲霉毒素B1相对合成标准不确定度。
由实验可知,试样中黄曲霉毒素B1的含量为15.70 μg/kg,取置信概率=95%,包含因子k=2,则试样中黄曲霉毒素B1的扩展不确定为UX=1.16 μg/kg。
试样中黄曲霉毒素Bl的测量结果为:(15.70±1.16) μg/kg,k=2。
2.8.4 主要不确定度来源的相对贡献
根据各来源的相对标准不确定度计算相应的所占百分比,评定相对贡献。由表13可知,标准储备液的不确定度贡献最大,其次是标准曲线配制和回收率。因此,在实验过程中高质量的标准物质、规范操作减小移液定容过程中产生的不确定度有助于降低检测结果的不确定度。此外,提高实验人员的实验技能及使用仪器设备的熟练操作、检测仪器的良好维护也有助于不确定度的减少。

表13 主要不确定度来源的相对贡献
3 结论
免疫亲和柱全自动固相萃取-超高效液相色谱法的加标回收率为 85%~104%,日内相对标准偏差为1.8%~3.0% ,日间精密度为1.8%~4.8%。测定三种基体质控物质的结果均在标示值范围内,测定结果准确可靠。通过配对t检验,仪器间无显著台间差,多次重复验证无仪器残留,方法稳定性优于手动法。利用本方法对试样中黄曲霉毒素B1测量不确定度进行了分析评定,不确定度的主要来源是标准储备液、标准曲线配制和回收率,在实验过程中高质量的标准物质、规范操作减小移液定容过程中产生的不确定度有助于降低检测结果的不确定度。本方法能够最大限度地减少实验人员的工作量,提高工作效率,还可避免因人员操作导致的结果偏差,适用于大量粮油中黄曲霉毒素的精准测定。
