4-苯基-2-(2-氯苯基)噁唑的合成及其荧光性质研究
2020-08-27胡青青荆补琴
解 海,胡青青,张 磊,李 吉,刘 文,荆补琴
(1.山西大同大学化学与环境工程学院,山西大同 037009;2.山西大同大学医学院,山西大同 037009)
噁唑环结构单元广泛存在于自然界、合成中间体、药物分子中,表现出良好的抗癌[1]、抗HIV-1[2]、抗过敏[3-6]等药理活性。如化合物A[2]是HIV-1 转录抑制剂,IC50值达0.03 μmol/L,可能成为一种抗病毒药物的先导化合物。噁唑衍生物还可以作为荧光探针及生物标记物[7-9],在生物或超分子体系中表现出很好的光学性质,如化合物B的苯并恶唑环中的氮和苯酚中氧原子与镁离子配位,形成平面配合物,这种共面性使荧光增强,使得化合物B可用于检测镁离子的荧光探针。见图1。

图1 含噁唑环结构单元的化合物A和B的化学结构式
Aza-wittig反应有原料易得、条件温和、操作简便的特点,已作为一种高效构建碳氮键的方法,在杂环合成的方法学研究中发挥了重要的作用。以前我们报道了一些通过烯基叠氮类化合物发生Aza-wittig反应合成各类杂环的新方法[10-14]。本研究利用简单容易合成的原料[15],通过发生连续的Staudinger 反应、分子内Aza-wittig 反应和杂环芳构化反应,一锅合成了4-苯基-4-(2-氯苯基)噁唑,该方法无需分离中间体、操作简单。
1 材料与仪器
1.1 材料
主要试剂有甲苯 (C6H5CH3)、二氯甲烷(CH2Cl2)、三乙胺(NEt3)、乙酰氯(CH3COCl)、石油醚,三苯基磷、无水硫酸钠,均为国产分析纯;甲苯、二氯甲烷用CaCl2干燥1 周使用,三乙胺(NEt3)重新蒸馏使用。测试荧光性质所用溶剂为色谱纯试剂,水为去离子水。
1.2 主要仪器
X-4型熔点仪(北京第三光学仪器厂),温度计未校正;PE-983 红外光谱仪(Perkin.Elemer 产品);PE-983 红外光谱仪(美国Perkin.Elemer 公司);Varian Mercury 600 型600 MHz 核磁共振仪(1H:600 MHz;13C:150 MHz,美国瓦里安有限公司);FinniganTrace 质谱仪(美国Thermo 公司);Vario EL III 元素分析仪(德国Elementar 公司);Scinco S-3100 UV-分光光度计(韩国Scinco 公司);FluoroMax-3-P 分光光度计(法国Jobin Yvon公司)。
2 实验过程
2.1 合成路线
反应以叠氮醇为起始原料,和2-氯苯甲酰氯在三乙胺为催化剂和缚酸剂的条件下反应,得中间体叠氮酯1,中间体1 再经三苯基磷处理,发生Staudinger反应得膦亚胺,该中间体无需分离,连续发生分子内Aza-wittig 反应和杂环芳构化反应,一锅合成2,4-二取代的噁唑。合成路线见图2。

图2 2-甲基-4-(44-氯苯基)-5-苯基噁唑的合成路线
2.2 化合物1的合成及表征
在100 mL圆底烧瓶中加入烯基叠氮醇(10 mmol,1.75 g),25 mL CH2Cl2作溶剂,保持反应温度在-5℃,向反应瓶中慢慢滴加重新蒸馏过的邻氯苯甲酰氯(10 mmol,0.75 g),之后在冰水浴条件下向反应瓶中以每秒2~3 滴的速度缓慢滴加三乙胺(15 mmol,2.1 mL),然后在冰水浴条件下继续搅拌反应0.5 h,撤掉冰水浴,在常温下继续搅拌,直至用薄层色谱(TLC)监测反应完成。将反应液缓慢倒入冰水中,用20 mL CH2Cl2萃取3 次,再用饱和食盐水洗涤萃取1次,萃取液用少量无水NaSO4干燥1h,抽滤掉NaSO4,在旋转蒸发仪上减压旋走CH2Cl2,暗红色残液经过硅胶短柱(洗脱液:V石油醚∶V乙醚=9∶1),后处理得到黄色固体叠氮酯1,质量为2.44 g,产率78%,熔点92~94 ℃,1HNMR(CDCl3,600 MHz):7.28~8.33(m,9H,Ar-H),6.04(s,1H,=CH),5.21(s,2H,CH2)。
2.3 化合物4的合成及表征
在100 mL圆底烧瓶中加入烯基叠氮酯(5 mmol,1.57 g),25 mL 甲苯溶解。在常温条件下向反应瓶中分批次加三苯基膦(5 mmol,1.31 g),常温下反应1.5 h,用薄层色谱(TLC)监测反应完成。不经过分离,将反应体系置于120 ℃油浴中回流搅拌6 h,TLC监测反应完毕,用旋转蒸发仪减压旋走甲苯溶剂,残液过硅胶短柱(洗脱液:V石油醚∶V乙醚=7∶1),得到2,4-二取代噁唑4,该物为红色固体,质量0.81 g,熔点103~104 ℃,产率65%。1HNMR(CDCl3,600 MHz):7.14~7.97(m,9H,Ar-H),6.77(s,1H,=CH),5.35 (s,2H,CH2);13CNMR(CDCl3,150MHz):32.8,123.5,123.9,126.6,126.9,128.5,128.7,129.4,132.8,136.3,137.8,142.9,148.3,159.4;MS:m/z(%)=269(48,M+),234(2),141(29),139(100),77(23)。C16H12ClNO 元素计算值:C,71.25;H,4.48;N,5.19;实测值C 71.46,H 4.64,N 4.96。
3 结果与讨论
合成的中间体和目标化合物均经过1HNMR、13CNMR、质谱和元素分析表征。通过解析中间体和目标化合物的1HNMR 谱图可知:芳氢的化学位移在7.19~8.33之间;关环后噁唑环中的氢原子受氧原子的影响,其化学位移在6.77,芳构化后特征的亚甲基的化学位移在5.35,为单峰。通过1HNMR 谱图,我们进一步验证了其发生芳构化的反应历程。通过解析4-苯基-2-(2-氯苯基)噁唑的13CNMR 谱图可知:亚甲基的特征峰在32.8;其他化合物的碳峰也可以找到应有的归属,得到合理的解释。EI-MS谱图中,合成的目标化合物的分子离子峰和碎片峰都能够得到合理的归属和正确的解释。
4-苯基-2-(2-氯苯基)噁唑为芳环取代的噁唑,存在共轭体系,噁唑和取代的苯环为刚性共平面结构,具有荧光性质。我们通过“参比法”测量了化合物3和4的荧光量子产率。
4-苯基-2-(2-氯苯基)噁唑的量子产率根据以下公式计算所得:
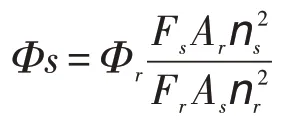
公式中Φ表示待测物的量子产率;F为荧光强度面积;A为激发波长处的最大吸光度值;n为溶液的折射率,下标的s和r分别代表标准物质和待测样品(公式计算要求:激发波长处的最大吸光度A不大于0.05)。
选择标准物质苯酚为参照,25 ℃时,其量子产率为0.14,化合物4 采用紫外定波长测量法,确定激发波长为378 nm,在该条件下,其量子产率计算得0.29。
通过荧光性质实验,我们发现4-苯基-2-(2-氯苯基)噁唑结构中,因噁唑环和苯环因间隔有一个亚甲基,噁唑环不能和苯环共平面,不能形成共轭体系,苯基对整个噁唑环的荧光发光基本没有贡献。而且,4-苯基-2-(2-氯苯基)噁唑中的另一个氯代苯环与噁唑环形成共轭体系,增大了噁唑环的共轭体系,对荧光强度有贡献。结果显示,4-苯基-2-(2-氯苯基)噁唑的荧光量子产率较参比物苯酚的大,其量子产率为基准物质苯酚的2倍左右。
4 结论
利用简单易得的原料,通过连续的Staudinger反应、分子内aza-wittig 反应和芳构化反应,合成了4-苯基-2-(2-氯苯基)噁唑。采用“参比法”研究了其荧光性能,其量子产率为0.29。通过改造噁唑环的取代基,进一步提高该类化合物的荧光量子产率,拓展其在荧光探针、生物标记以及在超分子体系中的应用。
