卡尔曼氏综合征1例报告
2020-08-17朱慧敏许小津张美英
朱慧敏,许小津,熊 燕,张美英
(南昌大学a.研究生院医学部2017级; b.第二附属医院内分泌代谢科, 南昌 330006)
卡尔曼氏综合征(kallmann综合征)是伴有嗅觉减退或缺失的特发性低促性腺激素性性腺功能减退症(IHH),而IHH是指因先天性下丘脑促性腺激素释放激素(GnRH)分泌或功能障碍,导致垂体分泌促性腺激素减少,引起性腺功能不足的疾病。kallmann综合征发病率低,临床上较少见,南昌大学第二附属医院2019年7月9日收治1例kallmann综合征患者,现结合相关文献对其临床资料进行回顾性分析,报告如下。
1 临床资料
患者,女,17岁,因自幼嗅觉缺失,至今无第二性征发育入院。患者家属诉患者自幼便出现嗅觉消失,无法辨别气味,当时未予以重视。至今患者无第二性征发育,乳房未生长,无腋毛及阴毛生长,无月经来潮。曾于2019年5月至江西省妇幼保健院就诊,查促卵泡生成素、促黄体生成素、雌二醇低,颅脑及磁共振提示双侧嗅球、嗅束未见显影。现为进一步明确诊断于2019年7月9日入本院内分泌科。入院查体:营养一般,体型消瘦,神志清楚,甲状腺未触及肿大。心率103次·min-1,律齐,未闻及杂音,双肺呼吸音清,未闻及干湿性啰音。腹软,无压痛及反跳痛。生理反射正常,病理反射未引出。双下肢无水肿。专科查体:身高158 cm,体重41 kg,体重指数(BMI)16.42 kg·m-2,血压101/71mmHg(1 mmHg=0.133 kPa),无皮肤色素沉着,无视野缺损,耳廓外形正常,粗测听力正常,鼻腔无异常分泌物,无法识别酒精、醋和清水气味。无牙龈缺损,无唇腭裂。双乳平坦(乳房Tanner分期Ⅰ期),无腋毛生长,无阴毛生长(阴毛Tanner分期:P1期),幼稚外阴。
辅助检查(2019-05-06江西省妇幼保健院)。抗穆勒氏管激素(AMH)1.97 ng·mL-1。性腺激素:促卵泡生成素(FSH)0.69 U·L-1↓,促黄体生成素(LH)0.19 mU·mL-1↓,雌二醇(E2)14.2 pg·mL-1↓,泌乳素(PRL)8.47 ng·mL-1(5.18~26.53 ng·mL-1),睾酮17.62 ng·dL-1(10.83~56.94 ng·dL-1),孕酮<0.1 ng·mL-1↓。游离甲状腺激素及抗体:FT3 3.72 pg·mL-1↑,FT4 0.91 ng·dL-1,TSH 5.27 mU·L-1↑,TgAb 493.52 U·mL-1↑,TPOAb 402.68 U·mL-1↑。妇科彩超:始基子宫?卵巢未探及。染色体分析:46,xx,320~400条带阶段G显带染色体分析未见明显异常。2019-06-05双能骨密度:正位腰椎Z评分-1.26,骨量减少。垂体/颅脑磁共振平扫:1)垂体MR未见异常;2)双侧嗅球、嗅束未见显影,符合Kallmann综合征表现(影像学资料丢失)。
入院后完善相关检查。1)一般检查:血常规、肝肾功能、电解质、尿粪常规、血糖、血脂、叶酸维生素B12、肿瘤四项等均正常。2)性激素检查:LH<0.07 mU·mL-1↓,FSH 0.98 mU·mL-1↓,E2 19.01 pg·mL-1↓,孕酮<0.21 ng·mL-1↓,PRL 15.32 ng·mL-1,睾酮28 ng·dL-1。3)GnRH兴奋试验:注射戈那瑞林25 μg前后各时点FSH、LH水平变化见表1。4)其他相关激素:游离甲状腺激素及抗体:FT3 3.83 pg·mL-1,FT4 1.05 ng·dL-1,TSH 10.617 mU·L-1↑,TgAb 425 U·mL-1↑,TPOAb>1300 U·mL-1↑。生长激素1.04 ng·mL-1。皮质醇(8:00)18.08 μg·dL-1,(0:00)1.15 μg·dL-1。醛固酮(卧位)10.39 ng·dL-1,(立位)27.31 ng·dL-1。血管紧张素(卧位)111.9 pg·mL-1↑,(立位)123.9 pg·mL-1↑,ACTH正常。5)影像学检查:常规心电图:窦性心律、房性早搏。双肾上腺CT平扫描+增强:左侧肾上腺内侧支稍增粗,增强后强化均匀。6)基因检测:患者FGFR1基因杂合缺失,其父母均阴性。
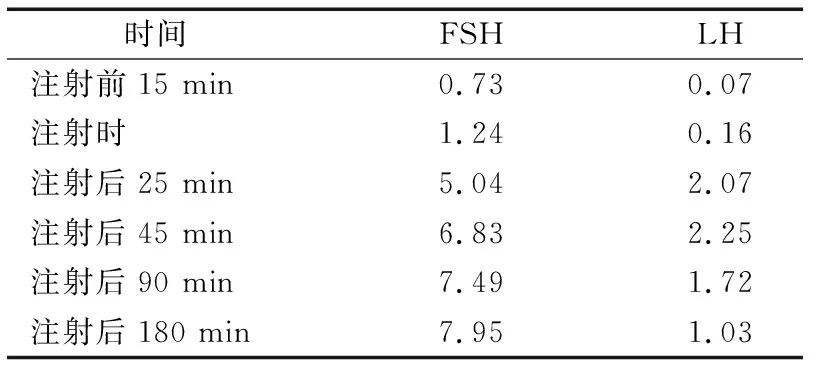
表1 注射戈那瑞林前后不同时刻患者激素水平情况 mU·mL-1
2 讨论
kallmann综合征是伴有嗅觉减退或缺失的低促性腺激素性性腺功能减退症,嗅觉异常这一特征将KS与其他类型的IHH区别开来,后者嗅觉功能正常。IHH临床少见,有研究[1]表明,IHH男性发病率为1/4000~1/10 000,女性发病率较男性低2~5倍;而Kallmann综合征约占IHH的40%~60%。Kallmann综合征是一种遗传异质性疾病,与遗传和基因突变有关,可呈常染色体显性或隐形遗传。随着基因组测序及生物信息学的发展,目前已报道有超过20个基因突变与Kallmann综合征有关,常见的包括:KAL1、FGFR1、FGF8、CHD7,ANOS1、HS6ST1、Sox10、SEMA3A、WDR11、IL17RD、PROKR2、PROK2,FEZF139[2-3]。这些基因在胚胎发育过程中参与一组专门处理嗅觉的神经细胞(嗅觉神经元)的形成和迁移,这些神经细胞起源于嗅觉区域的中枢神经系统之外,胚胎发育过程中迁移到大脑中嗅球的位置,而嗅球对嗅觉的感知起着至关重要的作用。GnRH神经元是起源于神经嵴和外胚层祖细胞,然后在发育过程中同嗅觉神经细胞一同迁移到大脑,这一途径就提供了嗅觉与生殖之间的发育联系。这些基因突变扰乱了嗅觉神经细胞和产生GnRH神经细胞的迁移,产生GnRH的神经元在大脑中的错位就无法产生GnRH,导致IHH的发生,此外,嗅觉神经细胞没有延伸到嗅球,这个个体的嗅觉就会受损或缺失,从而导致Kallmann综合征[4]。
有报道[5]提出,Kallmann综合征除嗅觉缺失外,也有腭裂和唇裂,少牙龈和手足裂,色盲,单侧肾发育不全,中枢听力障碍,手的镜像运动(同步运动)和共济失调,色盲。本例患者是一名青少年女性,临床上罕见,主要表现为嗅觉缺失及无第二性征发育。通过GnRH兴奋试验,发现LH及FSH基础值极低(其中LH低于0.7 mU·mL-1),予以戈那瑞林25 μg静脉注射后,LH峰值小于6 mU·mL-1,FSH值虽呈翻倍增长,但峰值延迟。结合患者垂体及颅脑磁共振结果报嗅球、嗅束无显影,可诊断患者Kallmann综合征。后进一步完善基因检测,发现FGFR1杂合缺失,该基因通常以常染色体显性遗传为主,但其父母均无该基因及其他相关基因的缺失,考虑患者FGFR1杂合缺失为基因突变所致,故进一步诊断明确。
Kallmann综合征女性患者在治疗上,通常采用雌孕激素替代治疗,该患者无第二性征发育,首先应予以戊酸雌二醇1 mg·d-1(6~12月),后增加剂量至2 mg·d-1(6~12月),补充雌激素,促进乳房、子宫、卵巢及外生殖器的发育,到达成人水平后,再周期性联合孕激素模拟正常生理周期,若要怀孕,则用脉冲式GnRH治疗,诱导规律月经及促进排卵[6]。同时,该患者伴有亚临床甲状腺机能减退及桥本甲状腺炎,需加用左甲状腺素钠片50 μg·d-1治疗。
Kallmann综合征作为一种特殊类型的IHH,发病率低,临床上罕见,很多患者对自己第二性征未发育未引起重视,或羞于启齿,不愿来院就诊,导致不能得到及时有效的治疗。同时临床上很多医师对该病不清楚或在诊治方面存在疑惑,造成漏诊或误诊,延误患者治疗。本案例,从临床表现、诊断到治疗,比较全面地展现了Kallmann综合征的诊疗思维,为临床医师提供了一个较好的病例。
