金银花总有机酸有效部位的制备及质量标准研究
2020-07-23王腾腾苏培文李孟孟李海刚
张 蕊,王腾腾,苏培文,李孟孟,黄 鹏,李海刚
(临沂大学,山东 临沂 276000)
有机酸类化合物是金银花的主要有效成分[1-2],包括绿原酸、异绿原酸、咖啡酸等有机酸类成分,具有抑菌抗病毒[3-4]、解热[5]、抗炎、保肝、止血、抗氧化、免疫调节等作用[6]。近年来人们对金银花绿原酸的提取方法[7-8]、金银花有机酸类成分的含量测定方法进行了广泛的研究,但将金银花总有机酸作为有效部位进行分离提取的报道尚不多见。本文在前期研究的基础上制备了金银花总有机酸有效部位,研究其薄层鉴别方法、总有机酸含量测定方法,采用高效液相色谱法测定了金银花总有机酸有效部位中绿原酸、异绿原酸A、异绿原酸B及异绿原酸C的含量,为金银花总有机酸有效部位的质量标准研究提供依据,为金银花总有机酸有效部位制剂的开发提供参考。
1 仪器与试药
1.1 仪器
1260型高效液相色谱仪(美国 Agilent公司);TU-1810spc紫外可见分光光度计(北京普析通用仪器有限公司);旋转蒸发仪(RE-2000A上海亚荣生化仪器厂);ME104E电子分析天平(梅特勒-托利多仪器有限公司)。
1.2 试药
绿原酸对照品(上海源叶生物科技有限公司,批号:Y24J7K16726);异绿原酸A对照品(中国食品药品检定研究院,批号:P28O7F23862);异绿原酸B对照品(中国食品药品检定研究院,批号P25J6F1793);异绿原酸C对照品(中国食品药品检定研究院,批号:P28D4S1);金银花(山东平邑);甲醇、乙腈为色谱纯,水为纯净水,其余试剂均为分析纯。
2 方法与结果
2.1 金银花总有机酸有效部位的制备
取金银花切段,加入10倍量70%乙醇回流煎煮3次,每次1h,滤过,合并3次煎煮液,滤过,取续滤液减压浓缩至相对密度1.2,石油醚萃取除去脂溶性成分,然后用等量乙酸乙酯萃取5次,弃去乙酸乙酯层,取水层上大孔吸附树脂LSA-7,径高比9∶1,先用4倍量柱体积水洗脱,洗脱流速3 mL/min,再用7倍量70%乙醇洗脱,洗脱流速5 mL/min,收集乙醇洗脱液,减压浓缩至干,即得金银花总有机酸有效部位,其中总有机酸含量约为71.6%。
2.2 薄层色谱鉴别
取金银花有机酸有效部位粉末50 mg置于10 mL量瓶中,加75%乙醇使溶解,作为供试品溶液。量取绿原酸对照品,加75%乙醇制成每1 mL含1 mg的溶液。照薄层色谱法(中国药典2015年版通则0502)试验,分别吸取上述两种溶液各10 μL,点于同一硅胶G薄层板上,以乙酸乙酯∶水∶乙酸∶二甲苯(85∶10∶20∶4)溶液为展开剂,饱和20 min后展开,取出,晾干,于365nm波长处观察,供试品色谱图中,在与对照品色谱相应的位置上,显相同颜色的斑点。结果见图1。
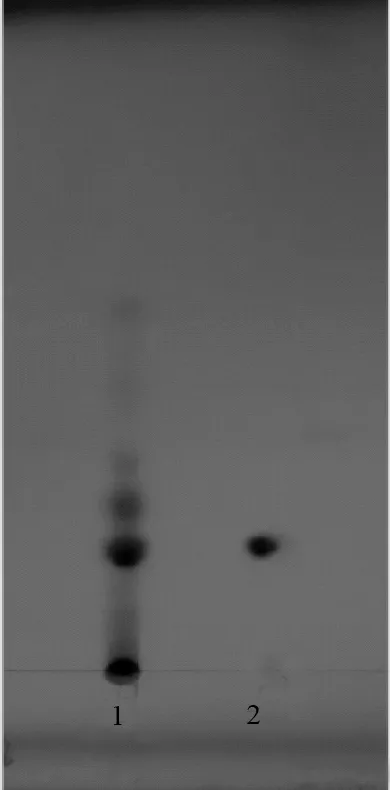
1.金银花有机酸有效部位;2.绿原酸
2.3 总有机酸含量测定
2.3.1 绿原酸对照品溶液配制 精密称取绿原酸对照品0.010 8 g,置于50 mL棕色量瓶中,加70%乙醇溶解并定容至刻度,摇匀,即得质量浓度为0.216 mg/mL的对照品溶液(10 ℃以下避光保存)。
2.3.2 最大吸收波长选择 取绿原酸对照品溶液适量,加70%乙醇稀释至适当浓度,按照紫外分光光度法于200~600 nm波长范围内进行紫外扫描,记录扫描色谱图,结果如图2所示。取金银花总有机酸提取物适量,70%无水乙醇溶解并稀释至适当浓度,同法于200~600 nm波长处扫描,记录扫描色谱图,结果如图3所示。
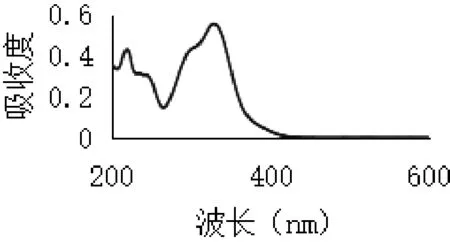
图2 绿原酸紫外扫描
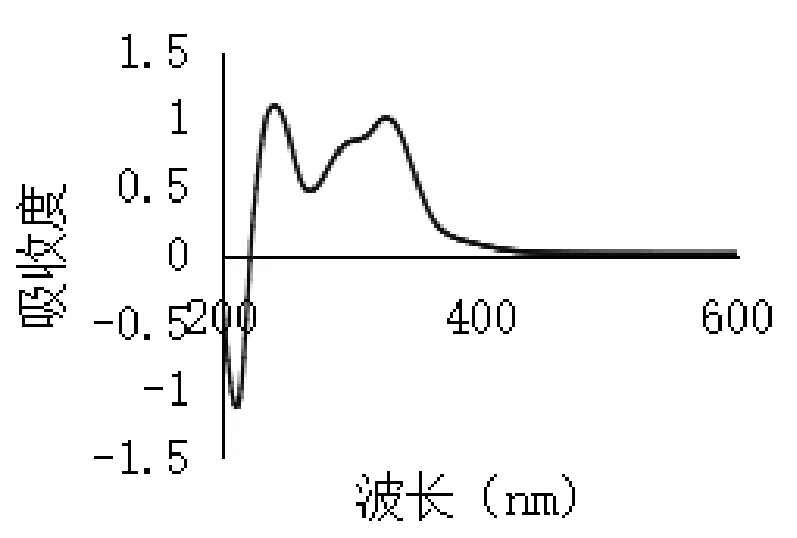
图3 总有机酸有效部位紫外扫描
由图可见,绿原酸在327 nm波长处有一最大吸收,总有机酸在243 nm及327 nm波长处有最大吸收,参考山东省药材标准[9](金银花提取物中总有机酸测定方法),结合本实验结果,本文选择327 nm作为吸收波长测定总有机酸含量。
2.3.3 标准曲线制备 精密量取绿原酸对照品溶液0.2 mL、0.4 mL、0.6 mL、0.8 mL、1.0 mL,置于10 mL容量瓶中,用70%乙醇稀释至刻度,以70%乙醇作空白对照,在327 nm处测吸光度。以质量浓度(x)为横坐标,吸光度(y)为纵坐标,绘制标准曲线,绿原酸在4.32~21.6 μg/mL浓度范围内与其吸光度线性关系良好,回归方程为y=48.936x+0.0425(R2=0.9994)。
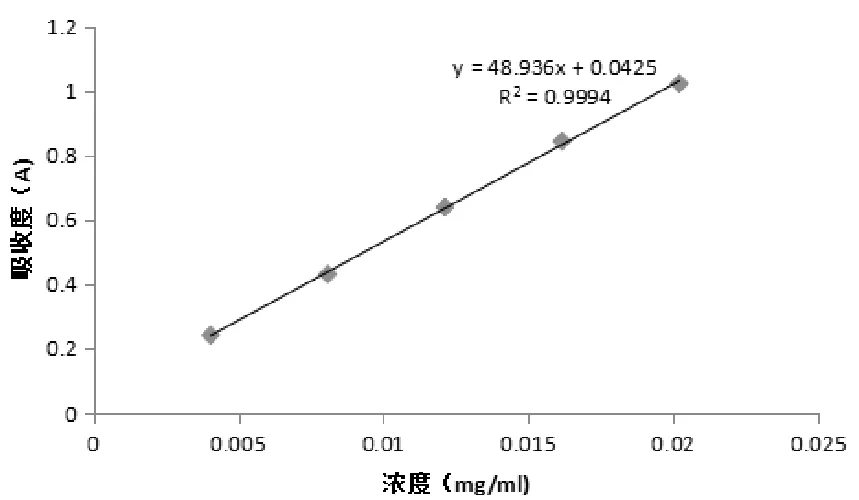
图4 绿原酸标准曲线
2.3.4 稳定性试验 精密量取绿原酸对照品溶液0.6 mL置于10 mL棕色容量瓶中,用70%乙醇稀释至刻度,摇匀,在0、2、4、6、8 h,于327 nm下测定吸光度。RSD为0.98%,溶液吸光度在8h内基本稳定。
2.3.5 精密度试验 精密量取绿原酸对照品溶液0.6 mL置于10 mL棕色容量瓶中,用70%乙醇稀释至刻度,摇匀,于327 nm下测定吸光度,连续测定6次,RSD为0.56%,表明试验所用仪器精密度良好。
2.3.6 样品回收率试验 分别精密量取绿原酸对照品溶液0.6 mL 9份,分别置于9个25 mL量瓶中,各量瓶均加入已知浓度的供试品溶液0.4、0.5、0.6 mL,70%乙醇稀释至刻度,摇匀,于327 nm下测定吸光度,计算总有机酸加样回收率,结果见表1,RSD为1.93%,说明该方法准确度良好。
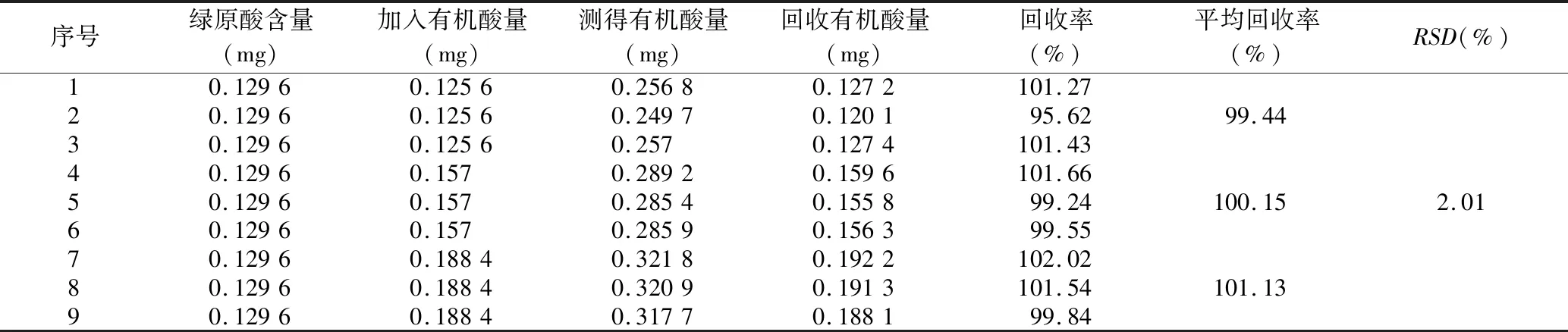
表1 总有机酸的加样回收率试验结果
参考山东省中药材标准中“金银花提取物”总绿原酸含量测定方法[9],结合本实验做如下变动,测定样品中总绿原酸含量,具体测定方法如下:精密称取绿原酸对照品0.010 8 g,置于50 mL棕色量瓶中,加70%乙醇溶解并定容至刻度,摇匀,即得质量浓度为0.216 mg/mL的对照品溶液(10 ℃以下避光保存)。取提取物粉末置于25 mL棕色量瓶中,加70%乙醇超声溶解并稀释至刻度,摇匀、滤过得供试品溶液。按照紫外分光光度法于327 nm波长处测定吸收度,以绿原酸为对照计算总有机酸含量,结果见表2。
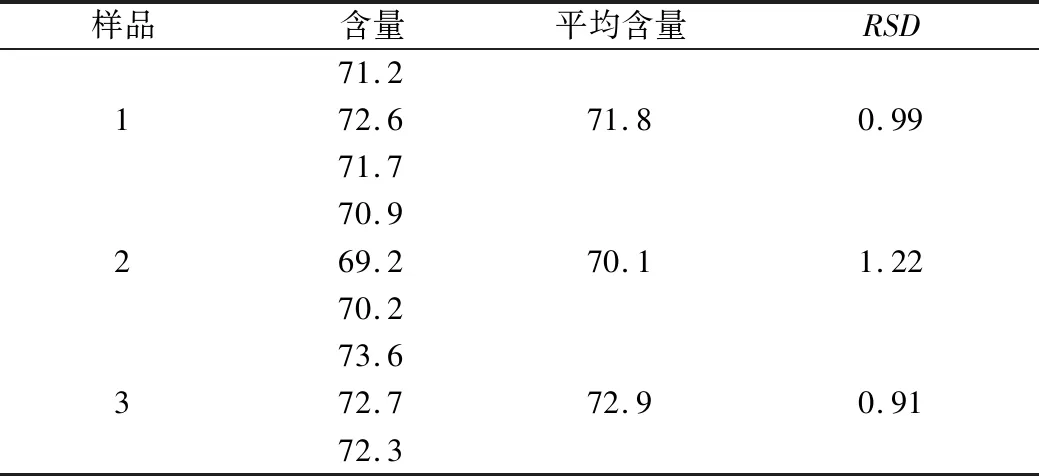
表2 金银花有机酸有效部位中总有机酸含量测定结果(%)
2.4 样品中指标成分含量测定
以绿原酸、异绿原酸A、异绿原酸B、异绿原酸C作为指标性成分,采用高效液相色谱法测定各指标性成分含量。
2.4.1 色谱条件 以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相A,以0.4%磷酸溶液为流动相B,梯度洗脱:0~15 min,10%~20%(A);15~40 min,20%~30%(A);40~60 min,30%~60%(A);检测波长为327 nm;柱温:室温;流速为1 mL·min-1;进样量为10 μL。
2.4.2 混合标准品溶液配制 分别精密称取绿原酸0.010 1 g、异绿原酸A 0.010 0 g、异绿原酸B 0.010 5 g、异绿原酸C 0.010 1 g,分别置于10 mL量瓶中,加50%甲醇溶解并稀释至刻度,得各指标性成分储备液。分别取各指标性成分储备液2.5 mL置于25 mL量瓶中,加50%甲醇溶解并稀释至刻度,得各指标性成分混合标准品溶液。
2.4.3 系统适应性 取混合标准品溶液,按照“2.4.1”项下色谱条件测定,绿原酸、异绿原酸B、异绿原酸A、异绿原酸C分离度良好,相邻色谱峰分离度均大于1.5,理论塔板数按绿原酸计算不低于2 000。
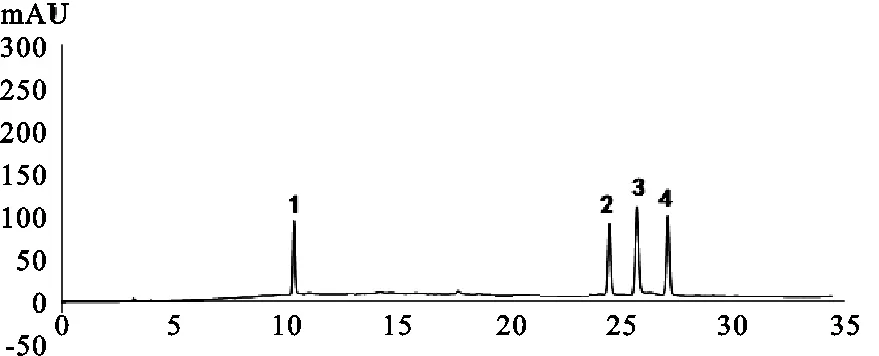
1.绿原酸;2.异绿原酸B;3.异绿原酸A;4.异绿原酸C
2.4.4 标准曲线制备 分别精密吸取“2.4.2”项下混合标准品溶液0.1、1.0、3.0、5.0 mL,置于10 mL量瓶中,70%乙醇稀释至刻度,摇匀,得系列浓度的混合标准品溶液,分别吸取10 μL注入液相色谱仪,记录色谱图,以峰面积对浓度进行线性回归,得回归方程,各对照品回归方程见表3。可见4种成分在0.01~1.0 mg·mL-1浓度范围内与其峰面积积分线性关系良好。
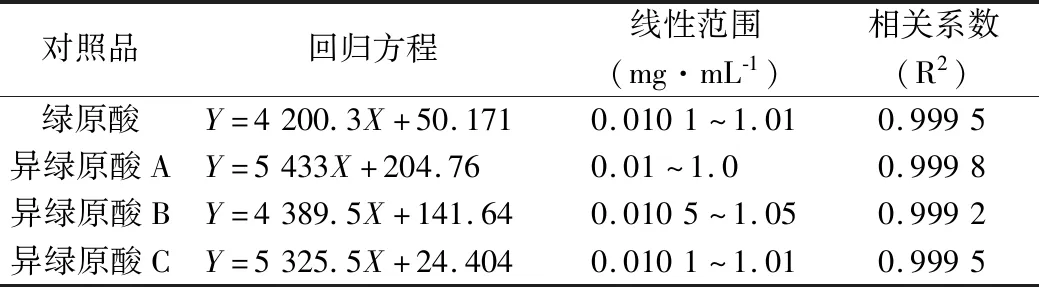
表3 4种对照品的回归分析结果
2.4.5 精密度试验 精密吸取混合对照溶液1.0 mL置于10 mL量瓶中,加70%的乙醇稀释至刻度,摇匀,分别取10 μL连续进样6次,结果绿原酸、异绿原酸A、异绿原酸B、异绿原酸C峰面积RSD分别为0.62%、0.77%、0.82%。
2.4.6 稳定性试验 精密吸取混合对照溶液1.0 mL置于10 mL量瓶中,加70%的乙醇稀释至刻度,摇匀,分别于0、2、4、6、8、12 h取10 μL进样分析,结果绿原酸、异绿原酸A、异绿原酸B、异绿原酸C峰面积RSD分别为0.72%、0.85%、0.94%。表明在12 h以内各对照品溶液稳定性良好。
2.4.7 重复性试验 精密吸取混合对照溶液1.0 mL 6份,分别置于10 mL量瓶中,加70%的乙醇稀释至刻度,摇匀,分别取10 μL进样分析,结果绿原酸、异绿原酸A、异绿原酸B、异绿原酸C峰面积RSD分别为0.51%、0.65%、0.74%,表明重复性良好。
2.4.8 加样回收率试验 分别精密称取已知含量的同一样品3份若干置于100mL量瓶中,分别精密加入各对照品溶液适量,加70%的乙醇稀释至刻度,摇匀,精密称取10 μL注入高效液相色谱仪,计算3种成分的加样回收率。由表4结果可以看出,在该色谱条件下,该定量方法对3种成分的加样回收率良好。
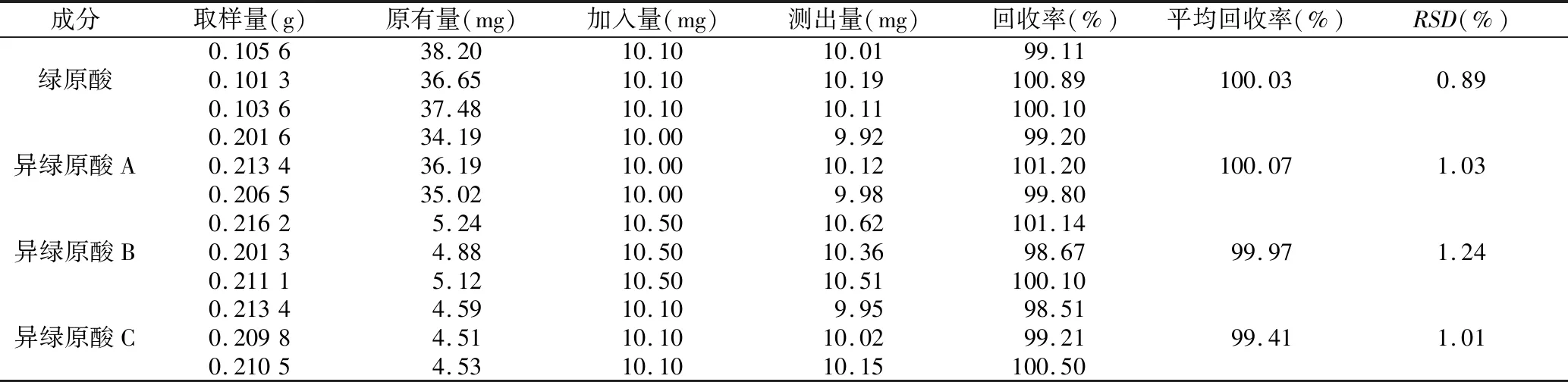
表4 4种成分的加样回收率试验结果
2.4.9 样品中绿原酸、异绿原酸B、异绿原酸A、异绿原酸C含量测定 取有机酸提取物样品置于100 mL量瓶中,加70%乙醇超声溶解并稀释至刻度,取10 μL注入液相色谱仪,记录色谱,按外标法计算样品中4种成分含量。见表5。

表5 金银花有机酸有效部位中4种成分含量测定结果
3 讨论
有机酸有效部位薄层鉴别图谱中绿原酸斑点清晰,其他成分斑点不够清晰,可能是含量较低的原因,因此在薄层鉴别中我们选择绿原酸作为有机酸有效部位薄层鉴别指标性成分。
流动相筛选过程中,考察了乙腈-水、甲醇-水、乙腈-磷酸、甲醇-磷酸、乙腈-醋酸、甲醇-醋酸梯度洗脱,发现以乙腈-磷酸为流动相时色谱峰峰形较好,分离度较高。绿原酸对照品溶液在200~800 nm全波长扫描,结果表明绿原酸在327 nm波长处有最大吸收。总有机酸提取物的200~600 nm全波长扫描结果显示出两个吸收峰(327 nm、243 nm),参照山东省中药材标准中“金银花提取物”总绿原酸含量测定方法测定样品中总绿原酸含量,选择327 nm作为检测波长。
