基于高通量测序分析青贮玉米微生物多样性
2020-07-04朱见深胡明齐静骆延波张庆李璐璐张印刘玉庆
朱见深 胡明 齐静 骆延波 张庆 李璐璐 张印 刘玉庆
摘要:青贮饲料发酵质量好坏很大程度上由其微生物组成决定。为了解青贮玉米微生物的多样性,利用高通量测序技术结合传统的纯培养技术,对31个青贮饲料样本进行微生物多样性分析。结果表明,青贮玉米微生物群落组成的共享核心菌属41个,占总样本微生物组成的87.7%,丰度较大的(≥1%)有10个菌属,其中乳杆菌属、克雷伯菌属及魏斯氏菌属分别占42.9%、12.1%、10.3%;真菌主要有念珠菌属、黑粉菌属、半乳糖霉菌属等。比较乳熟期组与蜡熟期组青贮的微生物群落,发现玉米的成熟度不同,青贮发酵产品中某些属微生物在数量上存在差别,但微生物群落组成无明显差异。本研究可为青贮饲料微生物群落结构及功能的研究提供参考。
关键词:青贮玉米;高通量测序;微生物多样性
中图分类号:S816.5+30.3文献标识号:A文章编号:1001-4942(2020)04-0068-05
Abstract The quality of silage fermentation is largely determined by its microbial composition. In order to understand the microbial diversity of maize silage, 31 silage samples were analyzed by high throughput sequencing technology combined with the traditional pure culture. The results showed that there were 41 shared core bacteria genera in silage maize microbial community, accounting for 87.7% of the total sample microbial community. Ten genera among them were more abundance than or equal to 1%, including Lactobacillus(42.9%), Klebsiella (12.1%), Weissella (10.3%) and so on. Candida, Ustilago and Galactomyces were the main genera of fugus. Comparing the microbial communities of silage samples using milk-ripe and ripening maize plants, it was found that there was no significant difference in the composition of microbial communities, but some differences in the number of microorganisms in some genera in silage fermentation products. This study could provide references for the researches of structure and function of microbial communities in silage.
Keywords Maize silage; High-throughput sequencing; Microbial diversity
青贮饲料(silage)是把新鲜的青饲料如玉米秸秆、根茎类植物以及牧草等,经碎化、包装、压紧、密封等处理后,进行微生物发酵处理,使青饲料中的纤维素降解并降低pH值,杀灭有害微生物,最大化地保留青饲料的营养成分及延长保存时间。青贮质量的好坏直接影响到反刍动物的生产性能[1]。
青贮过程是一个复杂的微生物发酵过程,有乳杆菌、芽孢杆菌、酵母菌、霉菌、腐败细菌等多种微生物参与,故青贮饲料品质的好坏与它所含的微生物有很大关系[2]。传统的菌群研究方法,如研究菌体及菌落的形态特征、生理生化特征、代谢产物等指标,一直沿用至今,但这些传统方法离不开纯培养技术,操作繁琐、复杂,耗费大量的时间和劳力;同时自然界的微生物在微环境中有各自的生态位,独立出来的细菌往往因失去菌群联合代谢网络很难在实验室条件下被纯化出来[3]。这些因素造成纯培养研究的结果不稳定,导致不能全面、客观地反映菌群结构与功能的真实情况。即使通过纯培养方法分离出优势菌株[4,5],再根据优势菌制备的青贮菌添加剂也不一定是青贮饲料的最佳佐剂[6]。
核酸测序技术的快速发展降低了测序成本,使高通量测序成为可能[7,8]。高通量测序近些年被大量地用于肠道菌群、海洋微生物等环境微生物的研究[9,10],也初步用于青贮微生物的研究[11,12]。本研究针对玉米植株成熟的不同階段,利用高通量测序技术结合传统纯培养技术对青贮玉米的微生物多样性进行分析,得到比传统纯培养方法更为全面的菌群数据,为青贮菌群的进一步研究提供参考。
1 材料与方法
1.1 样本收集
2019年4月于山东省德州市采集乳熟期(Ms,15份)和蜡熟期(Ds,16份)的全株青贮玉米材料,共31份,制成青贮料,不加任何添加剂,用封膜密封,进行自然无氧发酵。取样时,选取青贮玉米料内部无氧环境下的新鲜样品,放入抽净空气密封的无氧袋中,4℃冷藏,24 h内分析。
1.2 细菌、真菌培养及鉴定
MRS培养基分离纯化乳杆菌,PDA培养基分离纯化真菌,使用Omega DNA kit提取分离纯化菌株总DNA。分别进行16S rDNA与18S rDNA PCR扩增,产物由青岛擎科生物公司测序。将测序结果与NCBI数据库中的对应基因序列进行比对鉴定。其中,16S rDNA扩增引物为通用引物,序列为27F:5′-AGAGTTTGATCATGGCTCA-3′,1492R:5′-TACGGCTACCTTGTTAC-3′;18S rDNA扩增所用引物为基因特异性引物,序列为NS1:5′-GTAGTCATATGCTTGTCTC-3′,NS8:5′-TCCGCAGGTTCACCTACGGA-3′。
1.3 高通量测序试验方法
1.3.1 DNA抽提和PCR扩增 根据E.Z.N.A.soil试剂盒 (Omega Bio-tek, Norcross, GA, U.S.)说明书进行总DNA抽提,DNA浓度和纯度利用NanoDrop 2000进行检测,利用1%琼脂糖凝胶电泳检测DNA提取质量;用338F(5′-ACTCCTACGGGAGGCAGCAG-3′)和806R(5′-GGACTACHVGGGTWTCTAAT-3′)引物对V3—V4可变区进行PCR扩增(PCR仪:ABI GeneAmp 9700型)。扩增程序为:95℃预变性3 min;95℃变性30 s,55℃退火30 s, 72℃延伸30 s,27个循环;最后72℃延伸10 min。扩增体系为20 μL,包括: 5×FastPfu 缓冲液4 μL, 2.5 mmol/L dNTPs 2 μL, 引物(5 μmol/L)各0.8 μL, FastPfu聚合酶0.4 μL, DNA模板10 ng。
1.3.2 Illumina Miseq測序 使用2%琼脂糖凝胶回收PCR产物,利用AxyPrep DNA Gel Extraction Kit (Axygen Biosciences,Union City,CA,USA) 进行纯化,Tris-HCl洗脱,2%琼脂糖电泳检测。利用QuantiFluorTM-ST (Promega,USA) 进行检测定量。根据Illumina MiSeq 平台 (Illumina,San Diego,USA)标准操作规程将纯化后的扩增片段构建PE 2×300的文库。利用Illumina公司的Miseq PE300平台进行测序(上海美吉生物医药科技有限公司)。
文库构建步骤:(1)连接“Y”字形接头;(2)使用磁珠筛选去除接头自连片段;(3)利用PCR扩增进行文库模板的富集;(4)氢氧化钠变性,产生单链DNA片段。
1.3.3 数据处理 原始测序序列使用Trimmomatic 软件质控,使用FLASH软件进行拼接:设置50 bp的窗口,如果窗口内的平均质量值低于20,从窗口开始截去后端碱基,去除质控后长度低于50 bp的序列;barcode需精确匹配,引物允许2个碱基的错配,去除模糊碱基;根据重叠碱基overlap将两端序列进行拼接,overlap需大于10 bp。去除无法拼接的序列。
使用UPARSE软件(version 7.1 http://drive5.com/uparse/),根据97%的相似度对序列进行OTU聚类;使用UCHIME软件剔除嵌合体。利用RDP classifier (http://rdp.cme.msu.edu/) 对每条序列进行物种分类注释,比对Silva数据库(SSU123),设置比对阈值为70%。
2 结果与分析
2.1 青贮样品高通量测序OTU聚类
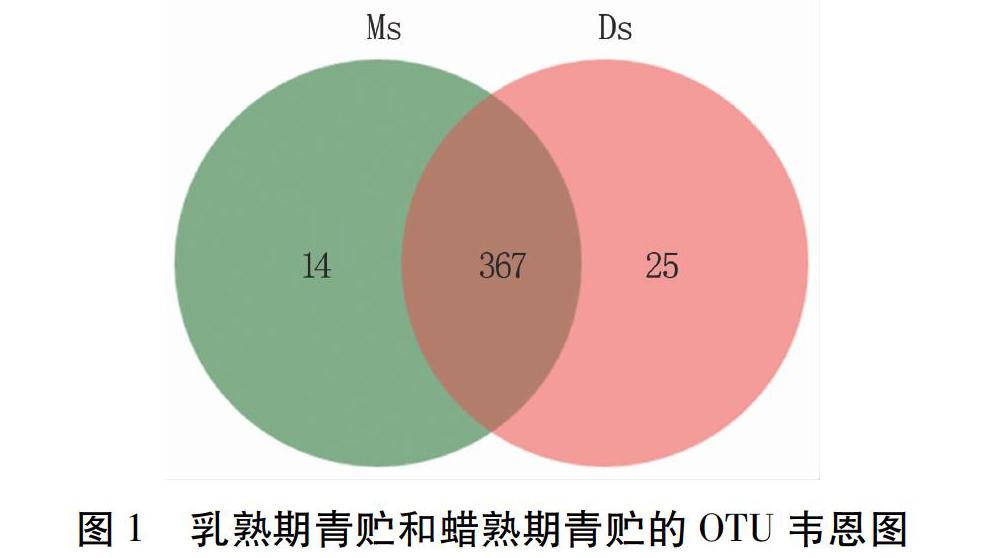
根据97%的相似度对序列进行OTU聚类后,总共获得406个不同的OTUs,其中乳熟期组(Ms)贡献381个,蜡熟期组(Ds)贡献392个,无显著差异。对两组做OTU韦恩图进行比较,发现乳熟期独有的OTUs个数为14个,蜡熟期独有的OTUs个数为25个,两组共享OTUs为367个。
2.2 不同成熟期玉米青贮的微生物多样性评价
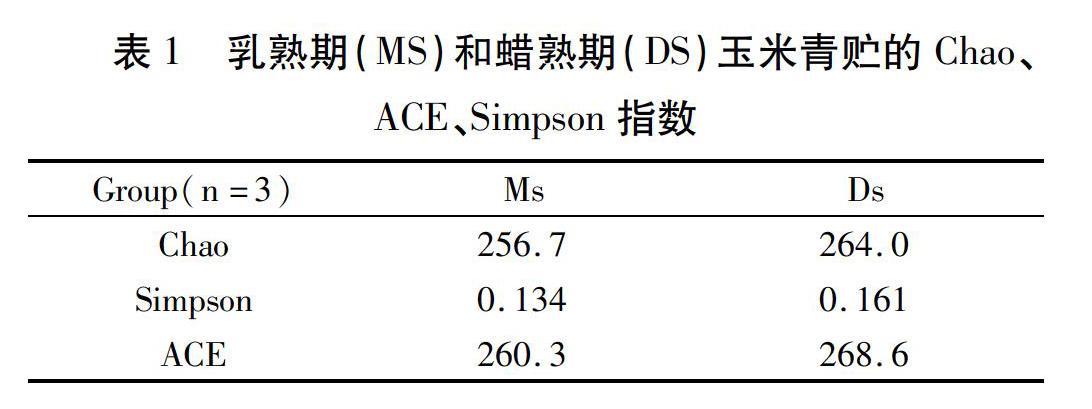
Chao是判断群落物种丰富度(abundance)的指数,值越大表示物种种类越多;ACE指数用于估算还有多少未被发现的物种,值越大,样本的真实物种种类越多;Simpson指数可用于综合评价样本中物种的丰富度与均匀度(evenness),其数值在0~1之间,当物种种类无限多(丰富度最高)且每个物种数目都一致(均匀度最高)时,Simpson值为1。由表1可以看出,蜡熟期组(Ds)较乳熟期组(Ms)多样性指数Chao、ACE大,说明蜡熟期组青贮玉米微生物多样性更高;且Simpson指数也较乳熟期组大,说明蜡熟期组的菌群组成丰富度和均匀度更高。
2.3 玉米青贮的微生物群落组成及丰度
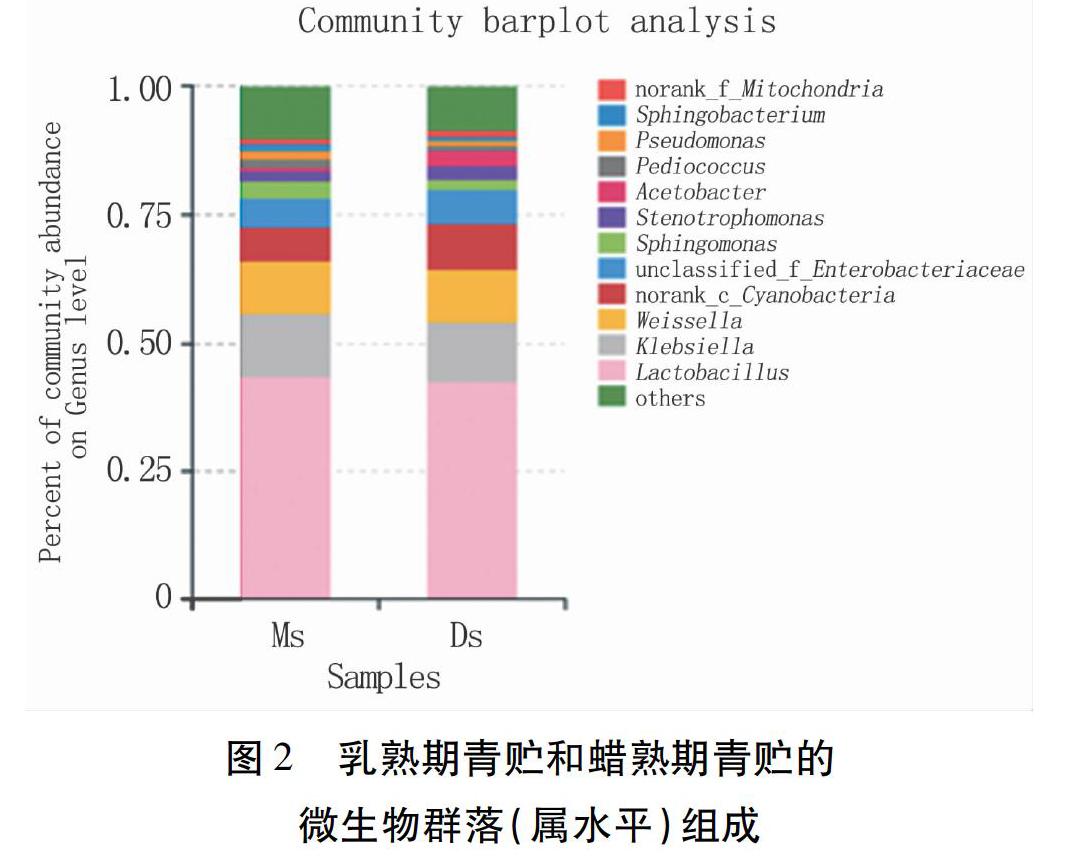
青贮玉米饲料厌氧发酵后,微生物群落主要由厚壁菌门(Firmicutes)、变形菌门(Proteobacteria)及蓝藻细菌类(Cyanobacteria)的微生物组成,其含量分别为55.55%、32.92%、8.58%。有41个共享核心菌属在所有样本中存在,占总样本微生物组成的87.7%。根据高通量测序结果在属水平上分析核心菌群,乳熟期和蜡熟期玉米青贮样品在菌群组成和含量上并无明显差异(图2),其中,乳杆菌属(Lactobacillus)是青贮发酵的主要菌属,占42.9%,其隶属于厚壁菌门芽孢杆菌纲乳杆菌目乳杆菌科,是导致样本中厚壁菌门微生物含量多的直接原因;其次为克雷伯菌属(Klebsiella)和魏斯氏菌属(Weissella),分别占12.1%和10.3%;三者为优势菌群,占总量的65%左右(表2)。除此以外,蜡熟期样品中蓝细菌(Cyanobacteria)、未分类的肠杆菌科(Enterobacteriaceae)和醋酸杆菌属(Acetobacter)略高于乳熟期样品,后者中鞘脂单胞菌属(Sphingomonas)则含量稍高。
使用MRS培养基分离优势乳杆菌并进行16S rDNA鉴定,获得的纯培养经鉴定为植物乳杆菌(Lactobacillus plantarum)、棒状乳杆菌(Lactobacillus coryniformis)、布氏乳杆菌(Lactobacillus buchneri)、干酪乳杆菌(Lactobacillus casei)等。
高质量的青贮依赖于无氧条件来降低pH值、抑制不利菌生长[13]。如果无氧条件不充分,则会使霉菌肆意生长,不仅造成青贮料营养成分流失,而且也会危及饲养动物健康[14]。本试验通过纯培养技术及18S rDNA鉴定,从青贮玉米料中分离到念珠菌属、黑粉菌属、半乳糖霉菌属、链格孢属及散囊菌属等真菌。
2.4 不同成熟期玉米青贮的菌群组成差异分析
经基于Bray_Curtis算法的OTU水平上的主坐标分析(PCoA),乳熟期与蜡熟期样品交错分布,但无显著聚类(图3)。表明,乳熟期和蜡熟期的玉米营养组成不同,青贮的微生物组成也存在些微差别,但差异并不显著。
3 讨论与结论
有研究表明乳杆菌属是青贮饲料菌群的关键发酵菌[15],本研究利用高通量测序及纯培养技术对玉米青贮的微生物多样性分析结果也印证了这点。乳杆菌为革兰氏阳性杆菌,菌体长形和细长,不产芽孢,通常无鞭毛,在无氧、酸性环境中生长,有时轻微需氧,CO2浓度在5%时能促进其生长,很多乳杆菌可选用MRS培养基进行分离纯化[16]。目前属内有185个种和亚种,代表菌种为德氏乳杆菌(Lactobacillus delbrueckii)[17]。伯杰手册介绍其多出现于植物表面,也存在于污水、动物肠道、阴道及口腔等环境中。在青贮中常见的乳杆菌有布氏乳杆菌(Lactobacillus buchneri)、植物乳桿菌、戊糖乳杆菌及干酪乳杆菌(Lactobacillus casei)等[18,19]。随着青贮发酵过程的深入,pH不断降低,较耐酸的乳杆菌属微生物更具优势,这可能是导致其含量最高的主要原因。
青贮质量的好坏主要依赖于青贮窖藏时的无氧环境,乳杆菌可以在无氧条件下发酵碳水化合物产生乳酸,降低pH,具有一定的抗菌能力,在添加48 h后迅速繁殖,无氧条件下经EMP途径发酵产生乳酸和乙酸[20]。本研究还发现乳熟期和蜡熟期两组发育成熟度不同的全株玉米用于青贮发酵,其微生物组成差异不明显,可以进一步结合青贮玉米营养成分等质量指标的分析,确定制作青贮的最佳生长阶段。
参 考 文 献:
[1] 刘艳丰, 唐淑珍, 巴音巴特, 等. 微生物青贮添加剂处理后玉米青贮对奶牛生产性能和经济效益的影响[J]. 中国畜牧杂志, 2012, 48(23): 76-78.
[2] 万学瑞, 吴建平, 雷赵民, 等. 优良抑菌活性乳酸菌对玉米青贮及有氧暴露期微生物数量和pH的影响[J]. 草业学报, 2016, 25(4): 204-211.
[3] Pande S, Kost C. Bacteria uncultureability and the formation of intercelluar metabolic networks[J]. Trends Microbiology, 2017, 25(5): 349-361.
[4] 李雁冰, 玉柱, 孙娟娟. 不同乳酸菌添加剂对青贮黑麦草和青贮玉米微生物群集的影响[J]. 草地学报, 2015, 23 (2): 387-393.
[5] 隋鸿园, 侯成立, 周雨霞. 适合作青贮接种剂植物乳杆菌的筛选[J]. 饲料研究, 2012(12): 68-69.
[6] Garrido-Cardenas J A,Manzano-Agugliaro F.The metagenomics worldwide [JP]research[J].Current Genetics, 2017, 63(5): 819-829.
[7] White R A, Callister S J, Moore R J. The past, present and future of microbiome analyses[J]. Nature Protocols, 2016, 11(11): 2049-2053.
[8] Degnan P H, Ochman H. Illumina-based analysis of microbial community diversity[J]. The ISME Journal, 2012, 6(1): 183-194.
[9] Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing[J]. Nature, 2010, 464(7285): 59-65.
[10]Hu Y, Yang X, Qin J, et al. Metagenome-wide analysis of antibiotic resistance genes in a large cohort of human gut microbiota[J]. Nature Communications, 2013, 4: 2151.
[11]熊乙, 赵燕梅, 许庆方, 等. 五个地区玉米青贮菌群多样性的研究[J]. 草学, 2017(5):16-22.
[12]闫绍鹏, 杨瑞华, 冷淑娇, 等. 高通量测序技术及其在农业科学研究的应用[J]. 中国农学通报, 2012, 28(30): 171-176.
[13]Yuan X F, Li P P, Wang H, et al. Enhancing the anaerobic digestion of corn stalks using composite microbial pretreatment[J]. Journal of Microbiology and Biotechnology, 2011, 21 (7): 746-752.
[14]李泽青, 闫峻, 张永芳, 等. 青贮发酵过程中主要微生物的活动规律及控制[J]. 广东畜牧兽医科技, 2011, 36(6): 12-15.
[15]陶莲, 刁其玉. 青贮发酵对玉米秸秆品质及菌群构成的影响[J]. 动物营养学报, 2016, 28(1): 198-207.
[16]陈东科,孙长贵. 实用临床微生物检测与图谱[M]. 北京:人民卫生出版社, 2011.
[17]党树锋, 侯俊财. 青贮饲料中乳酸菌的多样性分析[J]. 养殖技术顾问, 2011(10): 214-215.
[18]Ennahar S, Cai Y M, Fujita Y. Phylogenetic diversity of lactic acid bacteria associated with paddy rice silage as determined by 16S ribosomal DNA analysis[J]. Applied and Environmental Microbiology, 2003,69(1):444-451.
[19]王彦苏, 张一凡, 严振亚, 等. 水稻秸秆青贮饲料中可培养微生物多样性分析及优势乳酸菌的分离鉴定[J]. 草地学报, 2014, 22(3): 586-592.
[20]Kim J H, Block D E, Mills D A. Simultaneous consumption of pentose and hexose sugars: an optimal microbial phenotype for efficient fermentation of lignocelluosic biomass[J]. Applied Microbiology and Biotechnology, 2010, 88(5):1077-1085.
