金属负载变形石墨烯催化乙炔氯氢化反应机制
2020-07-04覃海川王丹阳李来才
覃海川,张 爽,王丹阳,王 薇,李来才
(四川师范大学化学与材料科学学院,四川成都610066)
聚氯乙烯是一种非常重要的工程塑料,它是由氯乙烯单体加工得来的,可以用作生产建筑材料、医疗器械、包装、服装材料等产品[1].当前工业生产氯乙烯主要有4种方法,分别是乙烯氧氯化法[2]、乙烷氧氯化法[3]、高温裂解法[1]和乙炔氢氯化法[4].由于中国的石油储蓄量稀少,煤矿资源却相对丰富,因此,我国主要采用乙炔氢氯化法生产氯乙烯[5].但是其催化剂 HgCl2挥发性强、毒性大,对人体健康和环境都有很大的危害,于是,对环境友好的、高性能无汞催化剂的研制与开发已经迫在眉睫[6].目前,在理论计算方面,文献[7]使用密度泛函理论(DFT)研究了金系催化剂的反应机制,反应分为2个阶段:1)乙炔和氯化氢吸附在氯化金上得到共吸附结构,2)氯化氢和共吸附结构继续反应得到氯乙烯和氯化金.赵璞君等[8]研究了CuCl2负载在石墨烯上催化C2H2和HCl反应生成氯乙烯的反应机制,该实验组探究了CuCl2作为活性组分在反应中不断失去活性的原因,以及石墨烯在掺杂N原子、P原子之后对催化C2H2和HCl反应生成氯乙烯反应的催化效果.在催化剂载体方面,乙炔氢氯化法的催化载体有活性炭载体、椰壳炭载体、氧化镁载体、氧化铝载体[9]、二氧化钛载体[10]、由氧化铝和二氧化钛组成的复合载体、二氧化硅载体[11]、分子筛载体[12]等.2013 年,Kawasumi等[13]合成出了一种新型材料,如图1所示,它们呈现出相互扭曲的形态,就像2个碗状的分子重叠错开放置,这种新型材料是一种石墨烯量子点(GQDs)C80H30.Dai等[14]利用 DFT的方法研究了该 GQDs的非线性光学性质,并预测了该GQDs是很好的非光学线性材料.到目前为止,在实验中,已经有许多人合成了含有多元环的GQDs,但是将变形石墨烯作为催化剂载体催化乙炔氢氯化反应的机理还没有人做过研究.本文使用变形石墨烯C80H30作为催化剂载体,研究了系列催化剂 PdCl2、HgCl2、MgCl2催化C2H2和HCl形成氯乙烯反应机制,通过比较不同路径的速控步骤的活化能大小,确定每种催化剂最优的反应路径.与之前的研究石墨烯负载二价金属催化剂催化C2H2和HCl进行比较,研究了催化反应活化能的变化,考察载体对该反应催化活性的影响特征.

图1 变形石墨烯晶体结构Fig.1 Deformed graphene crystal structure
1 计算模型和计算方法
把GQDs超晶胞用来模拟活性炭结构,使用了周期性的边界条件,并固定了下层碳原子,以1.600 nm的真空层把变形石墨烯晶体中上层碳原子充分优化.实验参数主要是GGA-PBE[15]描述的相关函数;使用Bergner等发明的ECP[16]对核电子进行描述;原子轨道基组用可极化的DNP[17]基组对其进行描述.将截断半径的大小设为0.370 nm.自洽迭代收敛的能量标准定为2.636×10-3kJ/mol.各能量的收敛标准设为2.636×10-2kJ/mol.使用LST/QST[18]方法寻找反应中的过渡态,同时使用频率分析和用 NEB[19]为基础的最小能量路径(MEP)验证所得到的过渡态的结构.
2 结果与讨论
研究了分别用 HgCl2、PdCl2、MgCl2作为催化剂,和氯化氢反应生成氯乙烯的反应机制.反应路径1、2、3的第一步都含二价金属化合物吸附在变石墨烯表面,二价金属 HgCl2、PdCl2、MgCl2吸附在变形石墨烯上的吸附能分别为 -75.7、-88.3、-63.6 kJ/mol,二价金属化合物吸附在变形石墨烯上的结构如图2所示.
如图3所示是路径1的反应流程图:第一步,二价金属催化剂(RCl2)吸附在变形石墨烯载体上;第二步,乙炔R1中的碳碳三键与RCl2中的R原子配位得到中间体M1;第三步,中间体M1中一个Cl原子靠近乙炔R1中的C原子,经由过渡态TS1逐步得到氯乙烯基汞(中间体M2);第四步,中间体M2上吸附HCl得到中间体 M3;最后,中间体 M3与HCl继续反应,其中H原子逐渐脱离HCl,Cl原子也逐渐脱离HCl经由过渡态TS2得到氯乙烯(产物 P).

图2 二价金属吸附在变形石墨烯上的结构Fig.2 Structure of bivalent metal adsorbed on deformed graphene
第一步研究了乙炔对催化剂 PdCl2、HgCl2、MgCl2中Cl元素的夺取,并优化了共吸附在graphite(001)表面的催化剂 PdCl2、HgCl2、MgCl2与乙炔.在表1中也给出了关键几何结构参数.如图2所示,催化剂 PdCl2、HgCl2、MgCl2与乙炔吸附于C=Cπ键.乙炔R1中的碳碳三键与二价催化剂PdCl2、HgCl2、MgCl2中的 Pd、Hg、Mg 原子配位生成中间体M1,中间体M1中的Cl(2)原子渐渐远离R原子,并逐渐靠近C(3)原子.经过过渡态TS1逐渐生成氯乙烯基汞(中间体M2).当催化剂为PdCl2、HgCl2、MgCl2时,经过频率分析,过渡态TS1只有一个负的频率,分别是 -261、-338、-257 cm-1.由表1可知,在催化剂分别为 PdCl2、HgCl2、MgCl2时,TS1 中 Cl(2)—R 的键长均被拉伸,而Cl(2)—C(3)的键长均被缩短.此步骤都需克服一定的能量,分别是23.9、101.7、74.1 kJ/mol.Cl原子夺取到的中间体M2,吸附于变形石墨烯表面.乙炔对催化剂PdCl2、HgCl2、MgCl2中 Cl元素的夺取具有一定的反应能量,分别为 42.7、72.4、62.8 kJ/mol.

图3 路径1的反应流程图Fig.3 The reaction flow chart in Scheme 1
第二步,研究了中间体M3和氯化氢继续反应,经过过渡态TS2得到氯乙烯的过程.如图3所示.氯化氢吸附在中间体M2上得到中间体M3.最后,氯化氢与中间体M3继续反应,其中氢原子渐渐脱离HCl,氯原子也逐步脱离HCl经由过渡态TS2得到氯乙烯(即产物P).在表1中提供了关键的几何结构参数.振动分析表明,当催化剂为 PdCl2、HgCl2、MgCl2时,经由频率分析,过渡态TS2只有一个负的频率,分别是 -215、-415、-289 cm-1.过程中H(1)原子渐渐远离 Cl(3)原子,并逐步靠近C(2)原子.Cl(3)原子逐渐远离 H(1)原子,并逐渐靠近R原子.由表1可知,在催化剂分别是PdCl2、HgCl2、MgCl2时,TS2 中 C(2)—R 的键长均被拉伸,Cl(3)—H(1)的键长均被拉伸.而 C(2)—H(1)的键长均被缩短.Cl(3)—R 的键长均被缩短.此过程皆需要克服一定的反应能,分别为42.7、129.3、53.6 kJ/mol.在催化剂分别是 PdCl2、HgCl2、MgCl2时,中间体M3经由过渡态TS2得到产物P的过程具有一定的反应能,分别为121.8、64.9、127.6 kJ/mol.
如图4所示为路径2的反应流程图,开始时二价金属催化剂RCl2吸附于变形石墨烯上,然后RCl2中的R原子和乙炔R1中的碳碳三键及HCl共同配位得到中间体Me,中间体Me中的氯原子和氢原子逐步靠近乙炔R1中的碳原子,经由过渡态TS逐步得到氯乙烯(产物P).
研究了 HCl中 H元素与 C2H2对催化剂PdCl2、HgCl2、MgCl2中 Cl元素的夺取过程.并优化了变形石墨烯表面反应物氯化氢、乙炔共吸附在催化剂 PdCl2、HgCl2、MgCl2上的结构.如图 4 所示,乙炔与催化剂 PdCl2、HgCl2、MgCl2及 HCl共吸附于C=Cπ键.二价催化剂 PdCl2、HgCl2、MgCl2中的Pd、Hg、Mg原子与乙炔R1中的碳碳三键及HCl共同配位得到中间体Me,中间体Me中的氯原子和氢原子逐步靠近乙炔R1中的碳原子,经由过渡态TS逐步得到氯乙烯(产物P).在表2中提供了关键的几何结构参数.由图可知,乙炔和催化剂 PdCl2、HgCl2、MgCl2及氯化氢吸附于C=Cπ键.当催化剂是 PdCl2、HgCl2、MgCl2时,经由频率分析,过渡态TS只有一个负的频率,分别是 -103、-185、-237 cm-1.其中Cl(1)原子逐渐远离 R原子,并逐渐靠近 C(3)原子.H(1)原子逐渐远离Cl(3)原子,并逐渐靠近C(3)原子.由表2可知,在催化剂分别是 PdCl2、HgCl2、MgCl2时,TS 中Cl(3)—H(1)的键长均被拉长,而 Cl(1)—C(2)的键长均被缩短,C(3)—H(1)的键长都被缩短.此过程皆需克服一定的反应能,分别为42.3、35.2、76.2 kJ/mol.乙炔对HCl中H元素和催化剂PdCl2、HgCl2、MgCl2中Cl元素的夺取步骤具有一定的反应能量,分别是 144.4、135.1、101.3 kJ/mol.
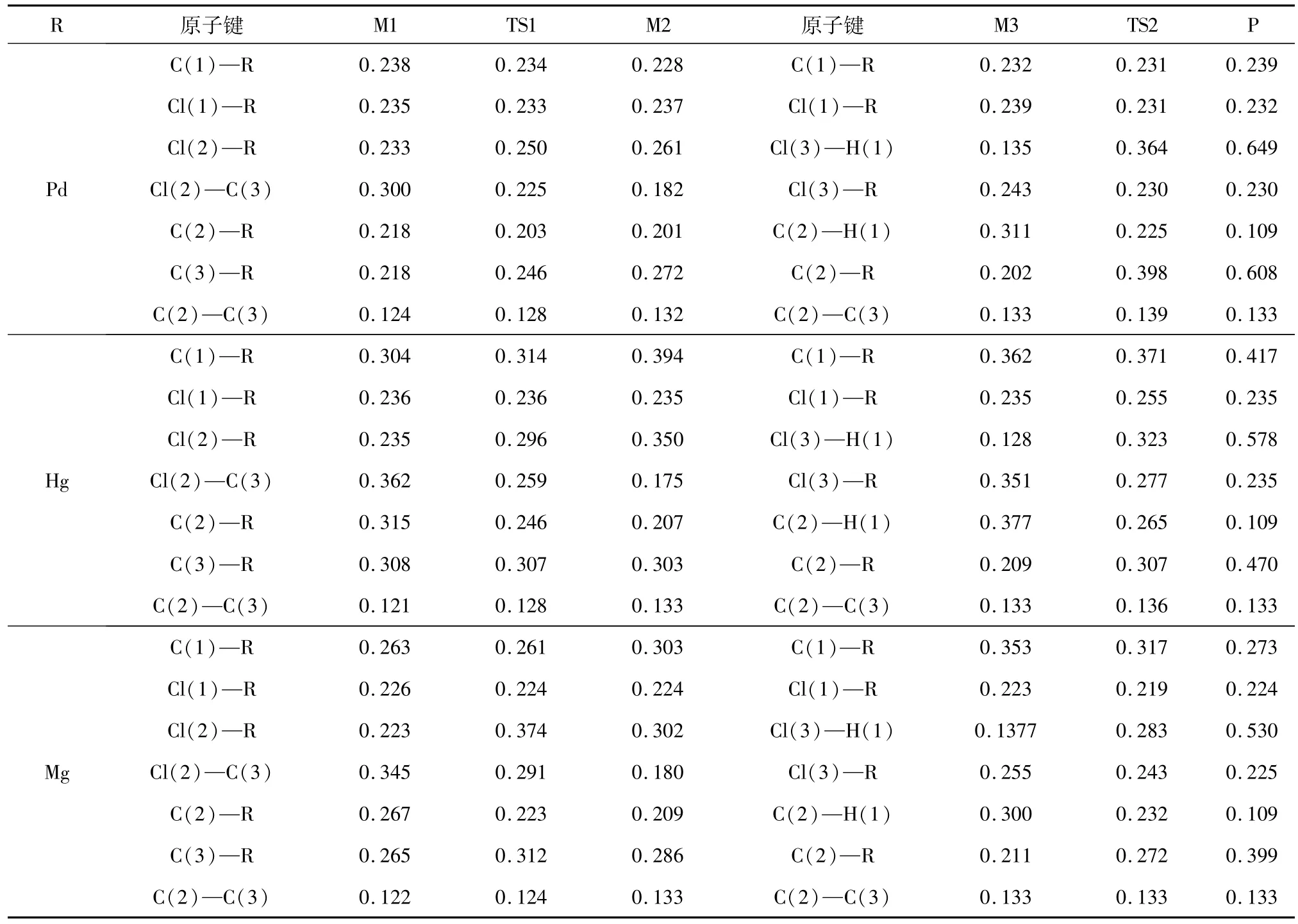
表1 路径1中各物质的键长变化表Tab.1 The bond length of each substance in Scheme 1 nm

图4 路径2中的反应流程图Fig.4 The reacting flow chart in Scheme 2
如图5所示是路径3的反应流程图,二价金属催化剂RCl2先吸附于变形石墨烯载体,然后RCl2中的R原子与乙炔R1中的碳碳三键及HCl配位得到中间体Me,中间体Me中的氢原子逐步靠近乙炔R1中的碳原子,经由TSA逐步得到中间体Me1.中间体Me1中氯原子逐步靠近乙炔R1中的碳原子,经由过渡态TSB得到氯乙烯(产物P).
最开始研究了C2H2对HCl中H元素的夺取过程.并优化了乙炔反应物氯化氢与催化剂PdCl2、HgCl2、MgCl2共吸附在 graphite(001)表面上的结构模型.如图5所示,在表3中也提供了关键的几何结构参数.二价催化剂 PdCl2、HgCl2、MgCl2中的Pd、Hg、Mg原子和乙炔R1中的碳碳三键及HCl配位得到中间体Me,中间体Me中的氢原子逐步靠近乙炔R1中的碳原子,经由 TSA逐步得到中间体Me1.当催化剂是 PdCl2、HgCl2、MgCl2时,经由频率分析,过渡态 TSA只有一个负的频率,分别是-201、-236、-216 cm-1.在这个过程中 H(1)原子渐渐远离 Cl(3)原子,并逐步靠近C(3)原子.由表3可知,在催化剂分别是PdCl2、HgCl2、MgCl2时,TSA中 Cl(3)—H(1)的键长均被拉伸,而Cl(3)—R的键长均增长,C(3)—H(1)的键长均被缩短.此过程皆要克服一定的反应能,分别为15.9、149.4、115.9 kJ/mol.乙炔对HCl中H元素的夺取步骤具有一定的反应能量,分别为54.4、147.3、105.0 kJ/mol.

表2 路径2中各物质的键长变化表Tab.2 The bond length of each substance in Scheme 2nm
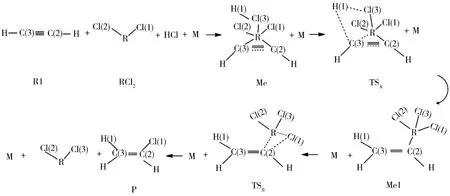
图5 路径3中的反应流程图Fig.5 The reaction flow chart in Scheme 3
接下来研究了乙炔对催化剂中Cl元素的夺取过程,如图5所示.在表4中也提供了关键的几何结构参数.中间体Me1中氯原子逐步靠近乙炔R1中的碳原子,经由过渡态TSB得到氯乙烯(产物P),其中Cl(2)原子逐渐远离R原子,并逐渐靠近C(2)原子.在催化剂是 PdCl2、HgCl2、MgCl2时,经由频率分析,过渡态 TSB只有一个负的频率,分别是-463、-358、-293 cm-1.由表3可知,当催化剂分别是 PdCl2、HgCl2、MgCl2时,TSB中 C(2)—R 的键长均被拉伸,而 Cl(1)—C(2)的键长均缩短.这个过程皆要克服一定的反应能,分别为28.0、43.5、8.4 kJ/mol.乙炔对催化剂中Cl元素的夺取步骤有一定的反应能量,分别是29.3、154.0、210.0 kJ/mol.

表3 路径3中各物质的键长变化表Tab.3 The bond length of substance in Scheme 3nm
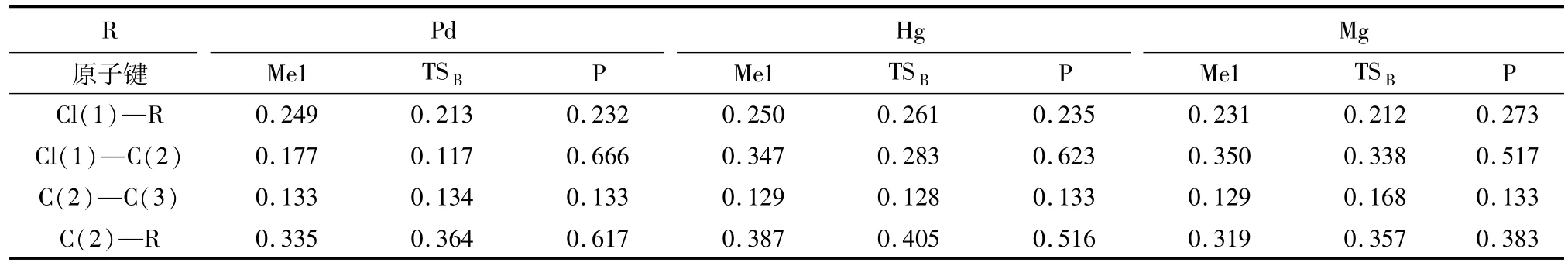
表4 路径3中各物质的键长变化表Tab.4 The bond length of each substance in Scheme 3nm
路径1、2、3中每个过程的活化能可由每个过程的相对能量所得,每个过程的相对能量如表5所示,每个过程的能级示意图如图6所示.

表5 各步骤的相对能量Tab.5 The relative energy of each step kJ/mol

图6 各步骤的反应能级图Fig.6 Schematic of energy levels
当催化剂为PdCl2时,每个反应路径的活化能分别是42.7、42.3、28.0 kJ/mol,反应都容易发生;其主要反应路径是路径3.催化剂为HgCl2时,各反应路径的活化能分别是129.3、35.2、149.4 kJ/mol,其主要反应路径是路径2.当催化剂为MgCl2时,各反应路径的活化能分别是74.1、76.1、115.9 kJ/mol,其主要反应路径是路径1.在路径1中,MgCl2的速控步骤是M1→TS1,PdCl2和HgCl2的速控步骤是M3→TS2;在路径2中,各催化剂的活化能都是Me→TS;在路径3中MgCl2和HgCl2的速控步骤都是Me→TSA.PdCl2的速控步骤是Me1→TSB.在最优反应途径中,PdCl2、HgCl2、MgCl2的反应活化能分别是28.0、35.2、74.1 kJ/mol.因此,反应所需活化能 PdCl2<HgCl2<MgCl2,所以催化剂性能PdCl2>HgCl2>MgCl2.与之前做的研究将石墨烯作为载体的结论一致,但是PdCl2所需的活化能增加了0.8、0.4、21.3 kJ/mol.这表明载体不同对催化剂催化乙炔氢氯化反应的催化活性也有一定的影响作用.
3 结论
本文使用了密度泛函的计算方法,以变形石墨烯(C80H30)作为催化剂载体,研究了二价金属系列催化剂(HgCl2、PdCl2、MgCl2)催化 C2H2和 HCl反应得到氯乙烯的微观反应机制.C2H2和HCl反应得到得到氯乙烯设计有三条不同的反应路径.根据每条反应路径的活化能大小,确定每种催化剂的最佳反应路径.再比较每种催化剂最佳的反应路径的活化能的大小,研究结果发现:各催化剂催化反应所需活化能大小MgCl2>HgCl2>PdCl2,故催化剂催化性能MgCl2<HgCl2<PdCl2,希望研究结果能为氯乙烯工业催化剂的使用提供理论参考.
