复方甘草片HPLC指纹图谱及7成分测定
2020-06-17高小惠胡燕琴王洪静刘军锋杨兆祥
高小惠,胡燕琴,王洪静,李 宁,2*,刘军锋*,杨兆祥
1昆药集团股份有限公司,昆明 650100;2上海中医药大学中药研究所教育部中药标准化重点实验室,上海 201203
复方甘草片是20世纪50年代我国医药工作者在国外经典天然药物处方的基础上,自主开发的镇咳祛痰药[1]。其原方出自约翰·布朗医师(1735~1788)的布朗合剂。1963年青海制药厂删除原方中酒石酸锑钾并改剂型为片剂,首家注册复方甘草片。此后复方甘草片的组方工艺几经变更[2]。目前处方每片含甘草浸膏粉112.5 mg、阿片粉或罂粟果提取物粉4 mg、樟脑2 mg、八角茴香油2 mg、苯甲酸钠2 mg[2,3]。复方甘草片治疗感染后咳嗽安全、可靠,总有效率为77.7%~85.4%,有效性及安全性与中华医学会呼吸病学分会《咳嗽的诊断与治疗指南(2015)》[4]推荐的右美沙芬相当[1],且较右美沙芬更为经济[5]。目前,中国有复方甘草片生产厂家33家,涉及36个批准文号,年产复方甘草片约200亿片,市场价值约20亿人民币[2]。
质量是药品疗效的根本保证。为提高仿制药质量,国务院办公厅2016年3月5日发布了《国务院办公厅关于开展仿制药质量和疗效一致性评价的意见》(国办发[2016]8号),要求开展仿制药质量和疗效的一致性评价。食品药品监管总局2016年5月25日发布了《2018年底前须完成仿制药一致性评价品种目录》,复方甘草片明确列入该目录中。2018年1月30日,仿制药质量与疗效一致性评价办公室将复方甘草片列入《289基药目录中国内特有品种名单》,鼓励并要求企业提出科学合理的评价方案。
为解决行业共性问题,进一步完善复方甘草片的质量研究,本研究收集10个厂家共18批次复方甘草片样品进行了摸底性调查研究,建立了HPLC指纹图谱分析方法,对其中14个共有峰进行了鉴定,并同时对其中吗啡、磷酸可待因、芹糖甘草苷、甘草苷、苯甲酸钠、异甘草苷、甘草酸7个主要成分的含量进行测定,为寻求符合复方甘草片特点的一致性评价路径提供新的技术手段和基础性研究资料。
1 材料、仪器与试剂
1.1 仪器
Agilent1200高效液相色谱仪(包括DAD检测器、柱温箱、自动进样器、在线脱气机、四元梯度泵)(美国Agilent公司);XP205、XPE26分析电子天平(Mettler公司);5510E-DTH超声提取仪(美国Branson公司);Milli-Q 超纯水机(美国Millipore公司)。
1.2 材料与试剂
18批复方甘草片(编号见表1)由昆药集团股份有限公司(以下简称昆药集团或昆药)提供,包括9批昆药集团样品和9批其他厂家(厂家1~9)样品;吗啡、磷酸可待因、苯甲酸钠、甘草苷、甘草酸铵、反式茴香脑对照品购自中国食品药品检定研究院;刺甘草查尔酮、甘草查尔酮A、甘草查尔酮B、甘草素、异甘草素、光甘草定、异甘草苷对照品购自成都曼思特生物科技有限公司;芹糖甘草苷对照品购自四川省维克奇生物科技有限公司;乙腈为色谱纯(MERCK公司),甲醇为分析纯(天津市风船化学试剂科技有限公司),水为超纯水,其他试剂均为分析纯。
2 方法
2.1 供试品溶液的制备
取本品20片,精密称取,研细,精密称取1片量,置50 mL量瓶中,加50%甲醇适量,超声提取30 min,用50%甲醇稀释至刻度,摇匀,滤过(0.45 μm滤膜),取续滤液,即得。
2.2 对照品溶液的制备
精密称取各对照品适量,加少量甲醇溶解后,用50%甲醇水溶液稀释定容,即得各对照品储备液。根据实验需要,分别精密吸取各储备液适量,用50%甲醇水溶液稀释定容配成混合对照品溶液。其中含量测定用吗啡、磷酸可待因、芹糖甘草苷、甘草苷、苯甲酸钠、异甘草苷、甘草酸铵混合对照品溶液,每 1 mL分别约含吗啡0.008 mg、磷酸可待因0.004 mg、芹糖甘草苷0.036 mg、甘草苷0.016 mg、苯甲酸钠0.040 mg、异甘草苷0.016 mg、甘草酸铵0.180 mg。
2.3 色谱条件
Welch Ultimate AQ-C18色谱柱(5 μm,4.6 mm×250 mm);以乙腈为流动相A,以0.02 mol/L磷酸盐缓冲液(取磷酸二氢钾2.72 g,加水至1 000 mL,磷酸调pH至4.0)为流动相B,进行梯度洗脱(0~10 min,2%→10% A;10~25 min,10%→38% A;25~35 min,38%→55% A;35~40 min,55%→60% A;40~50 min,65% A;50~55 min,65%→2% A;55~65 min,2% A);流速1.0 mL/min;检测波长为220和254 nm;柱温30 ℃;进样量10 μL。
3 结果
3.1 指纹图谱的方法学考察
3.1.1 精密度试验
取同一批号昆药集团样品,按“2.1”项下方法制备供试品溶液,按“2.3”项下的规定连续进样6次,测得各色谱图的相似度均大于0.99,RSD小于0.5%。以甘草酸色谱峰为参照,主要共有峰相对保留时间RSD小于1%,相对峰面积RSD小于3%。结果表明仪器精密度良好。
3.1.2 重复性试验
取同一批号昆药集团样品,按“2.1”项下方法平行制备6份供试品溶液,按“2.3”项下的规定进样分析,测得各色谱图的相似度均大于0.99,RSD小于0.5%。以甘草酸色谱峰为参照,主要共有峰相对保留时间RSD小于1%,相对峰面积RSD小于3%。结果表明本方法重复性良好。
3.1.3 稳定性试验
取同一批号昆药集团样品,按“2.1”项下方法制备供试品溶液,分别于制备后的0、2、4、8、12、18、24、48、72、96 h进样,按“2.3”项下的规定进样分析,测得各色谱图的相似度均大于0.99,RSD小于0.5%。以甘草酸色谱峰为参照,主要共有峰相对保留时间RSD小于1%,相对峰面积RSD小于3%。结果表明样品于96 h内稳定。
3.2 指纹图谱的建立、相似度分析及共有峰指认
取不同批号复方甘草片共18批,分别按“2.1”项下方法制备供试品溶液,按“2.3”项下的规定进样分析,将220 nm波长的色谱图输入相似度评价软件进行相似度比较,剪切10 min之前和50 min之后的色谱峰,采用中位数法生成参照指纹图谱。各批次药品指纹图谱的叠加图及共有模式指纹图谱分别见图1和图2。以共有模式为参照,各批次样品的相似度计算结果见表1,所有批次样品的相似度均在0.95 以上。指纹图谱中可见27个主要共有峰(图2),占总峰面积80%以上。与对照品色谱图对照保留时间,共有峰1、3、8、9、10、14、16、17、19、20、22、24、25、26分别指认鉴定为吗啡、磷酸可待因、芹糖甘草苷、甘草苷、苯甲酸钠、异甘草苷、甘草查尔酮B、甘草素、甘草酸、刺甘草查尔酮、异甘草素、甘草查尔酮A、光甘草定、反式茴香脑。

图1 18批样品指纹图谱Fig.1 Fingerprints of 18 batches of samples

图2 18批样品指纹图谱共有模式Fig.2 Fingerprint common patterns of 18 batches of samples
3.3 指纹图谱的相对保留时间和相对峰面积
以19号峰甘草酸为参照峰计算各共有峰的相对保留时间和相对峰面积,结果见表2。19号峰结构确切,与相邻色谱峰分离良好,易于识别,且保留时间及峰面积均较稳定,适于作为参照峰。共有峰的相对峰面积分布在一个较宽的范围内,最大RSD(26号峰)达61.66%,客观反映了化学成分的批间差异。
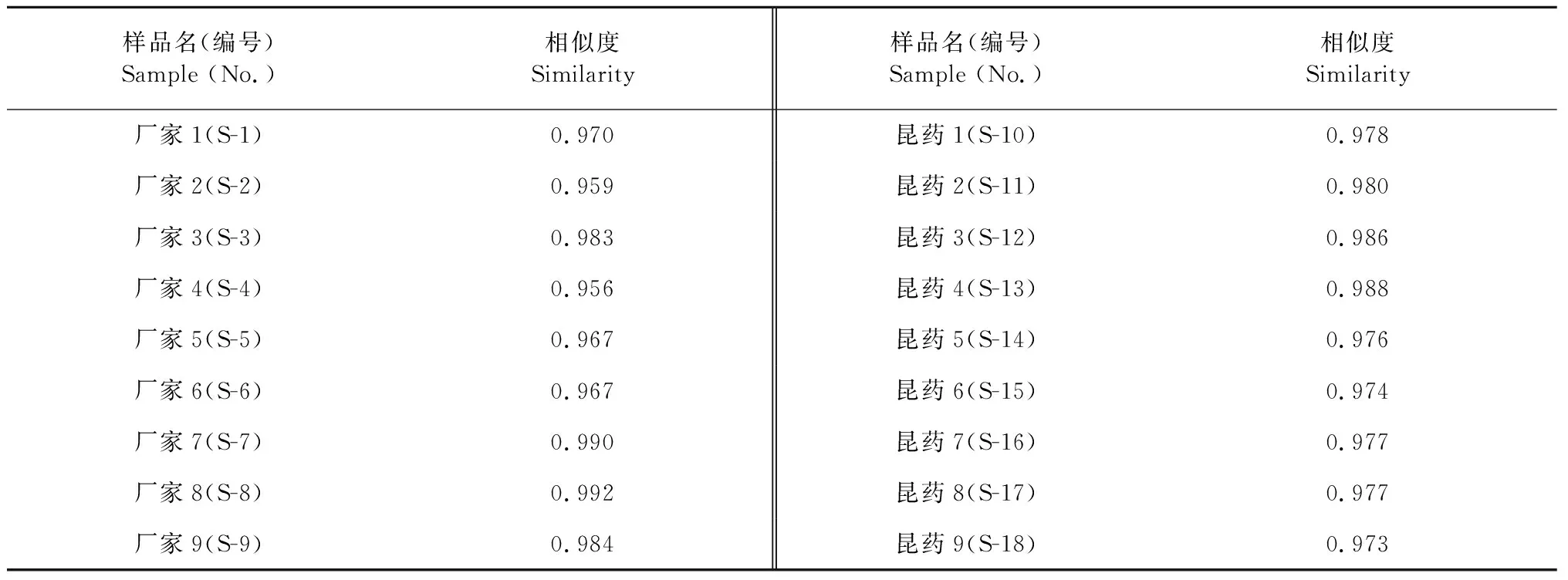
表1 18批样品相似度Table 1 Similarities of 18 batches of samples

表2 18批样品共有峰相对保留时间及相对峰面积Table 2 The relative retention time and relative peak areas of common peaks of 18 batches of samples
续表2(Continued Tab.2)
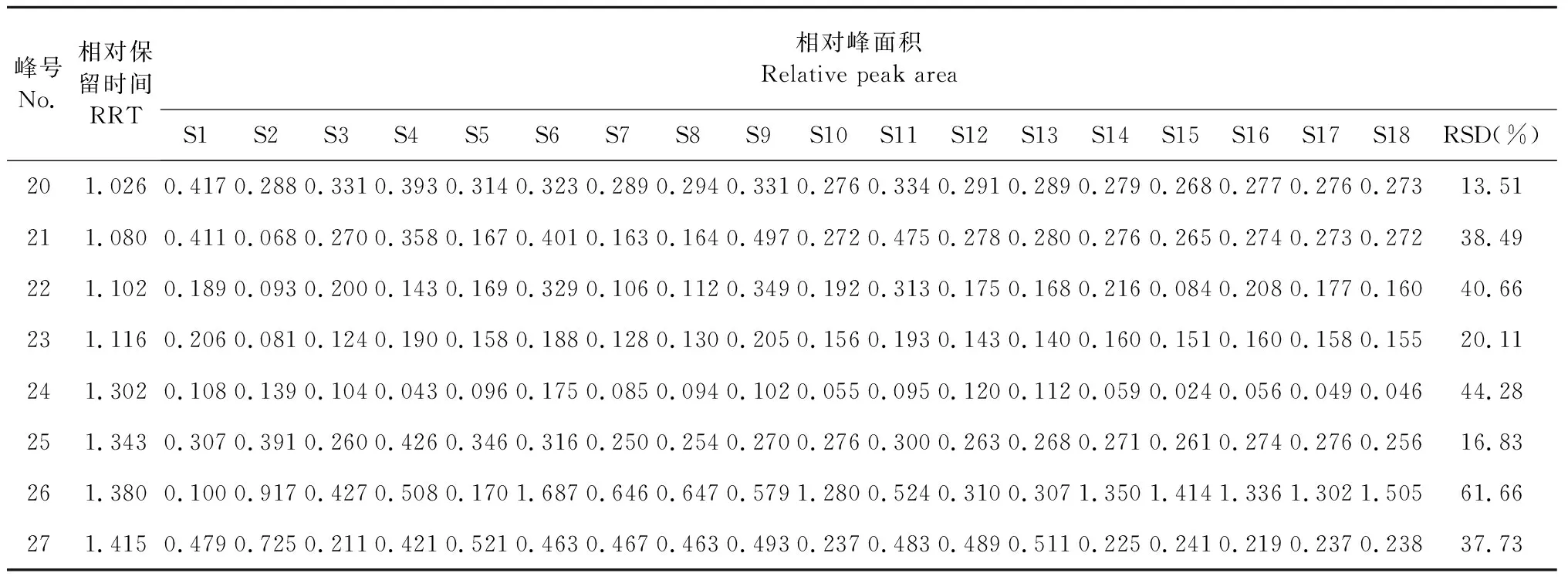
峰号No.相对保留时间RRT相对峰面积Relative peak areaS1S2S3S4S5S6S7S8S9S10S11S12S13S14S15S16S17S18RSD(%)201.0260.4170.2880.3310.3930.3140.3230.2890.2940.3310.2760.3340.2910.2890.2790.2680.2770.2760.27313.51211.0800.4110.0680.2700.3580.1670.4010.1630.1640.4970.2720.4750.2780.2800.2760.2650.2740.2730.27238.49221.1020.1890.0930.2000.1430.1690.3290.1060.1120.3490.1920.3130.1750.1680.2160.0840.2080.1770.16040.66231.1160.2060.0810.1240.1900.1580.1880.1280.1300.2050.1560.1930.1430.1400.1600.1510.1600.1580.15520.11241.3020.1080.1390.1040.0430.0960.1750.0850.0940.1020.0550.0950.1200.1120.0590.0240.0560.0490.04644.28251.3430.3070.3910.2600.4260.3460.3160.2500.2540.2700.2760.3000.2630.2680.2710.2610.2740.2760.25616.83261.3800.1000.9170.4270.5080.1701.6870.6460.6470.5791.2800.5240.3100.3071.3501.4141.3361.3021.50561.66271.4150.4790.7250.2110.4210.5210.4630.4670.4630.4930.2370.4830.4890.5110.2250.2410.2190.2370.23837.73
3.4 含量测定的方法学考察
本研究建立的指纹图谱分析方法可同时对复方甘草片中吗啡、磷酸可待因、芹糖甘草苷、甘草苷、苯甲酸钠、异甘草苷、甘草酸面积7个主要成分进行含量测定。
3.4.1 专属性试验
取昆药集团样品及空白辅料,按“2.1”项下方法制备供试品溶液和空白辅料溶液,取对照品,按“2.2”项下方法制备对照品溶液,分别按“2.3”项下的规定进样分析。如图4中可见,空白辅料溶液色谱图中无干扰峰影响定量,对照品溶液与供试品溶液色谱图中吗啡、磷酸可待因、芹糖甘草苷、甘草苷、苯甲酸钠、异甘草苷、甘草酸与相邻峰分离良好(分离度大于1.5)。结果表明本方法专属性良好。
3.4.2 精密度试验
取同一批号昆药集团样品,按“2.1”项下方法制备供试品溶液,按“2.3”项下的规定连续进样6次,测得吗啡、磷酸可待因、芹糖甘草苷、甘草苷、苯甲酸钠、异甘草苷、甘草酸峰面积RSD为0.68%~1.29%。结果表明仪器精密度良好。
3.4.3 重复性试验
取同一批号昆药集团样品,按“3.1”项下方法平行制备6份供试品溶液,按“2.3”项下的规定进样分析,测得吗啡、磷酸可待因、芹糖甘草苷、甘草苷、苯甲酸钠、异甘草苷、甘草酸峰面积RSD为0.21%~3.90%。结果表明本方法重复性良好。
3.4.4 稳定性试验
取同一批号昆药集团样品,按“2.1”项下方法制备供试品溶液,分别于制备后的0、2、4、8、12、18、24、48、72、96 h进样,按“2.3”项下的规定进样分析,测得吗啡、磷酸可待因、芹糖甘草苷、甘草苷、苯甲酸钠、异甘草苷、甘草酸的峰面积 RSD为0.98%~4.53%。结果表明样品于96 h内稳定。
3.4.5 线性关系试验
精密称取7个对照品,以50%甲醇水溶液溶解并逐级稀释成一系列不同浓度的工作液。按“2.3”项下的规定进样分析。于220 nm检测吗啡、磷酸可待因、芹糖甘草苷、甘草苷、苯甲酸钠和异甘草苷,254 nm检测甘草酸。以浓度为横坐标X,峰面积为纵坐标Y绘制工作曲线,计算回归方程。结果见表3,吗啡、磷酸可待因、芹糖甘草苷、甘草苷、苯甲酸钠、异甘草苷、甘草酸在相应的质量浓度范围内线性关系良好。
3.4.6 定量限
取各成分对照品溶液,逐级稀释测定,满足S/N≥10,即为定量限。结果表明吗啡、磷酸可待因、芹糖甘草苷、甘草苷、苯甲酸钠、异甘草苷、甘草酸的定量限为2.3、4.5、7.3、3.8、9.5、4.0、4.8 ng。
3.4.7 加样回收率试验
分别取已知含量的同一批号昆药集团样品9份,每份约0.0685 g置50 mL量瓶中,精密称取。分为3组,分别加入供试品中待测定成分量为80%、100%、120%的对照品。以上9份按“2.1”项下方法,自“加50%甲醇适量”开始制备加样回收供试品溶液,按“2.3”项下的规定进样分析,计算加样回收率。吗啡、磷酸可待因、芹糖甘草苷、甘草苷、苯甲酸钠、异甘草苷、甘草酸的平均加样回收率分别为104.45%、103.47%、100.07%、100.60%、97.54%、94.73%、102.83%,RSD值分别为0.81%、3.89%、0.68%、0.89%、2.91%、1.52%、1.35%。结果表明本方法回收率良好。
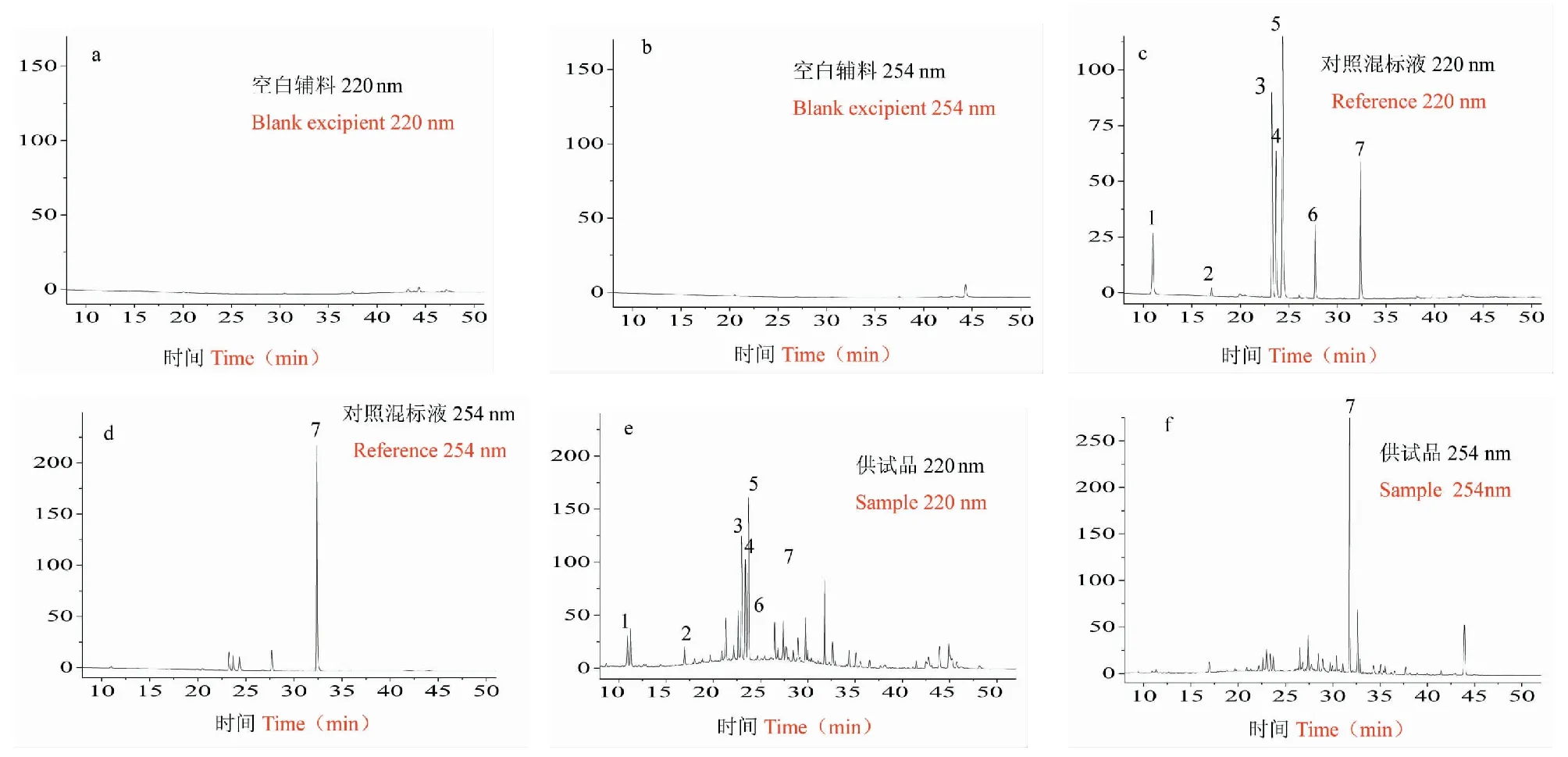
图3 空白(a和b)、对照品(c和d)和供试品(e和f)的色谱图Fig.3 Chromatograms of blank (a,b),reference (c,d) and sample (e,f)注:1.吗啡;2.磷酸可待因;3.芹糖甘草苷;4.甘草苷;5.苯甲酸钠;6.异甘草苷;7.甘草酸。Note:1.Morphine;2.Codeine phosphate;3.Liquiritinapioside;4.Liquiritin;5.Sodium benzoate;6.Isoliquiritin;7.glycyrrhizic acid.

表3 7种指标成分的线性关系Table 3 Linear relationships of 7 constituents
3.5 样品含量测定
取18批样品,按“2.1”项下方法制备供试品溶液,取吗啡、磷酸可待因、芹糖甘草苷、甘草苷、苯甲酸钠、异甘草苷、甘草酸对照品,按“2.2”项下方法制备对照品溶液,分别按“2.3”项下的规定进样分析,根据标准曲线计算各批次样品中7个化合物的含量,结果见表4。
4 讨论
复方甘草片作为复方天然药物,化学成分复杂,质量受药材来源及工艺参数等多种因素的影响,其一致性评价方法与化学仿制药有显著的区别。中药、天然药物的一致性评价是复杂的系统科学,是行业普遍关注的热点问题,众多学者提出了多种研究模式与思路[2,6-8]。其中,指纹图谱和多成分含量测定是较为公认的技术手段与评价方法。建立指纹图谱分析方法,与多成分含量测定相结合,构建中药整体成分质量控制体系,已成为中药质量标准的发展方向[9]。
复方甘草片的指纹图谱及含量测定方法已有相关文献报道[10-15]。但不同研究者的角度、研究目的及实验条件不同,报道的分析方法各异,多数无法在获得指纹图谱的同时进行多成分精确含量测定,且测定的指标性成分数量较少。本课题组前期采用UHPLC-LTQ-Orbitrap高分辨液质联用技术从复方甘草片中分析鉴定了55个成分[16]。该方法可用于进一步获取复方甘草片的化学轮廓及55个成分的定量与半定量。但方法测试成本较高且需要配备专门的仪器及分析人员。为解决行业共性问题,本研究基于HPLC及紫外检测器建立了更为通用的分析方法,更加适于工业界推广应用。

表4 18批样品7成分含量测定结果Table 4 Results of content determination of 7 constituents
本研究的色谱条件参考文献[13],并在此基础上进行了优化调整。首先,采用了耐水的反相色谱柱。为保证吗啡及相邻色谱峰的基线分离,本方法以98%水相做为起始流动相,耐水色谱填料的使用可避免高水相引起的色谱柱塌陷,延长色谱柱寿命。其次,选择220 nm为指纹图谱的检测波长。该波长下的色谱图色谱峰数量多,峰形及分离度良好,来源于甘草浸膏粉、阿片粉或罂粟果提取物、八角茴香油的色谱峰及苯甲酸钠均有体现,且各色谱峰峰面积比例适中。虽然254 nm波长的化学信息也较丰富,但甘草酸色谱峰的峰面积占总峰面积的比例过大,对整体相似度贡献过大。第三,选择220 nm做为吗啡、磷酸可待因、芹糖甘草苷、甘草苷、苯甲酸钠、异甘草苷、甘草酸的定量波长。选择254 nm做为甘草酸的定量波长。对7个成分进行了完整的含量测定方法学考察。其中吗啡、磷酸可待因为阿片粉或罂粟果提取物的活性成分,芹糖甘草苷、甘草苷、异甘草苷和甘草酸为甘草浸膏粉的特征性成分,且兼顾了苯甲酸钠,多指标共同定量使分析结果更具代表性。
本研究首先通过指纹图谱相似度评价法,通过与对照指纹图谱计算相似度评价药品质量。18批样品的相似度均在0.95以上,总体来说,不同厂家、批次复方甘草片的指纹图谱具有相对较好的一致性。其次对主要共有峰的相对峰面积范围进行了比较分析。相对峰面积反映了各色谱峰的比例关系,进而更加细致地反映制剂的内在质量。在本研究中,各批次药品共有峰的相对峰面积在较宽的范围内波动,客观反映了不同批次样品成分比例的差异。本研究进而对7个指标性成分进行了含量测定,18批制剂7成分含量总和的平均值超过处方总重量的10%。18批复方甘草片中,吗啡、可待因、苯甲酸钠、甘草酸的含量一致性较好,RSD分别为3.89%、6.20%、6.92%和6.79%。芹糖甘草苷、甘草苷、异甘草苷的含量一致性较差,RSD分别为15.86%、18.17%和14.34%。本研究对10个厂家18批次市场流通样品进行了分析,对复方甘草片的质量进行了初步的摸底性调查研究。但复方甘草片生产企业多,产量大,批次多,未来的工作中,建议利用本研究建立的分析方法,进一步增大样本数量,对不同厂家、批次样品的质量情况进行更加深入的调查研究。
阿片粉或罂粟果提取物粉属于国家管控的麻醉药品,由指定的企业统一供应,各复方甘草片厂家所用的原料来源相对一致,成品中吗啡和可待因的含量也相对稳定。甘草浸膏是甘草经加工制成的浸膏[3],生产厂家较多,来源多样。甘草为多基源药材,至少涉及甘草GlycyrrhizauralensisFisch.、胀果甘草G.inflataBat.、光果甘草G.glabraL.三种药用植物[3],物种本身的化学成分和活性存在差异[17],野生及栽培药材的质量参差不齐[9],不同品种、不同采收期栽培药材的有效成分含量亦有不同[18]。各厂家浸膏生产工艺的差异进一步造成了甘草浸膏粉的质量差异。复方甘草片现行国家标准[3]对甘草酸含量进行了规定,因此各厂家采取了技术手段以保障甘草酸含量的相对稳定。但是对于未订入质量标准的成分,如芹糖甘草苷、甘草苷、异甘草苷等,批间含量波动较大,一致性较差。在未来的研究工作中,应加强对药材源头的研究和管控,系统研究各成分在药材、中间体、制剂间的转移传递规律,制订更为科学、合理的质量标准。本研究为进一步研究提供了分析方法和技术保障。
5 结论
本研究建立了复方甘草片的HPLC指纹图谱及吗啡、磷酸可待因、芹糖甘草苷、甘草苷、苯甲酸钠、异甘草苷、甘草酸 7成分测定方法。实验结果表明本方法简便、快速、准确、可靠,可用于复方甘草片的质量控制,为寻求符合复方甘草片特点的一致性评价路径提供了新的技术手段和基础性研究资料。
