艾司氯胺酮与N-甲基-D-天冬氨酸受体的分子作用机制
2020-06-12张丽雷徐梦晓张静晓张文曦唐文强
张丽雷,徐梦晓,张静晓*,张文曦,唐文强
(1.洛阳师范学院 化学化工学院,河南 洛阳 471934;2.洛阳师范学院食品与药品学院,河南 洛阳 471934;3.陕西国际商贸学院医药学院,陕西 西安 712046)
抑郁症通常表现为情绪低落,睡眠质量差,自尊心低下,以及失去对娱乐活动的兴趣,并且具有高度复发的可能性。有研究表明重度抑郁症的遗传性为37%至48%[1],是严重影响人们正常生活的精神类疾病[2-4]。另外,抑郁症是一种多因素疾病,主要与遗传、生物学、环境和社会因素有关[5]。根据现代的心理学和生物学概念,抑郁症不仅是精神障碍,而且也是生理疾病,具有明确的生物学基础,例如重度抑郁症病人会出现脑神经损伤,神经可塑性下降和神经元回路异常等生理问题[6],需要使用药物进行治疗。
氯胺酮是N-甲基-D-天冬氨酸受体(NMDAR)的拮抗剂,由于能够快速改善抑郁症状和降低自杀意念,作为新型的抗抑郁药物受到了广泛的关注[7-9],但是由于具有致幻和滥用的风险限制了其临床应用。艾司氯胺酮是氯胺酮的对映异构体,与NMDAR具有更强的结合力,并同样具有改善抑郁症状和降低自杀意念的作用。然而,艾司氯胺酮抗抑郁的作用机制并不十分清楚,本文从艾司氯胺酮对NMDAR的拮抗机制出发,结合分子对接和分子动力学方法研究艾司氯胺酮与NMDAR的作用机制,为阐明艾司氯胺酮的抗抑郁机制和新型抗抑郁药物的开发提供理论依据。
1 材料与方法
从蛋白晶体结构数据库(RCSB PDB)中获取NMDAR的晶体结构(3OEM),去除N-甲基-D-天冬氨酸配体和结晶水,利用PDB2PQR软件包[10]补全缺失的原子,记为NMDAR,作为NMDAR的初始结构。从ATB数据库[11]中获取艾司氯胺酮的分子结构和力场数据(Molid:204037)。采用AutoDock Vina程序[12]对艾司氯胺酮和NMDAR进行对接,口袋大小设为20×20×20Å3,网格间距设定为1.0Å。
采用GROMACS(2018.4)软件包[13]进行分子动力学模拟,将分子对接得到的艾司氯胺酮与NMDAR的复合物作为分子模拟的初始结构,采用GROMOS 54A7力场。在初始结构周围延伸8Å的立方体中填充水分子,建立周期性结构,并加入2个氯离子使体系保持中性,最终整个体系大约包含60000个原子。首先对整个体系进行1000步的能量极小化动力学,再进行100ps的限制性动力学,使溶剂弛豫,之后再进行20ns的NPT分子动力学模拟。在模拟过程中,设置时间步长2fs,运用Velocity-rescale方法控制整个体系的温度为310K,采用parrinello-rahman方法控制体系的压力为1bar,使用PME方法计算长程静电相互作用,范德华截断值设为12Å。使用GROMACS软件包对分子模拟的结果进行分析,并采用VMD软件对结果进行可视化。
2 结果与讨论
艾司氯胺酮与NMDAR蛋白对接的结果如图1(a)所示,其中,艾司氯胺酮分子以球棍模型表示,NMDAR以蛋白的二级结构表示。从图中可见,艾司氯胺酮分子成功对接于蛋白C端(CT)叶片和N端(NT)叶片中间的口袋位置。考察了艾司氯胺酮的结合引起的NMDAR结构和运动趋势的变化,NMDAR对接前后模拟组合轨迹的主成分porcupine图如图1(b,c)所示,其中,箭头表示在蛋白质在第一振动模式下骨架原子的运动方向。从图中可见,对接之前的NMDAR蛋白的运动主要是C端和N端叶片结构在逆时针方向上的有序运动,其中NT的运动振幅更大,C端振幅较小,运动更加无序。当NMDAR与艾司氯胺酮对接之后,蛋白NT的运动改为顺时针的运动,并且运动趋势减弱,也更加无序。这可能是由于艾司氯胺酮的对接减少了有序刚体叶片的旋转运动程度,起到了稳定蛋白结构的作用。

图1 艾司氯胺酮与NMDAR对接后的结构(a),和NMDAR对接前(b)后(c)分子模拟运动的porcupine图Fig.1 Docking configuration of s-ketamine with NMDAR,and porcupine diagrams of the NMDAR before (b) and after (c) docking
为了确定分子模拟过程中蛋白和配体的稳定性,考察了模拟过程中蛋白和配体结构的均方根偏差(RMSD),结果如图2所示。从图中可见,经过约3ns的模拟之后,NMDAR对接前后的结构中蛋白和配体的RMSD值均趋于稳定,上下波动范围稳定在2Å范围之内,表明所研究结构在经过平衡后均达到稳定结构。NMDAR对接前后结构的RMSD没有发生明显的改变,表明艾司氯胺酮的对接对NMDAR的整体结构影响不大。另外,模拟过程中艾司氯胺酮的RMSD值变化较小,表明艾司氯胺酮的结构和位置没有发生明显的变化,结合稳定。

图2 NMDAR与艾司氯胺酮对接前后分子模拟过程中的RMSD图Fig.2 RMSD of NMDAR before and after docking withs-ketamineduring molecular simulation
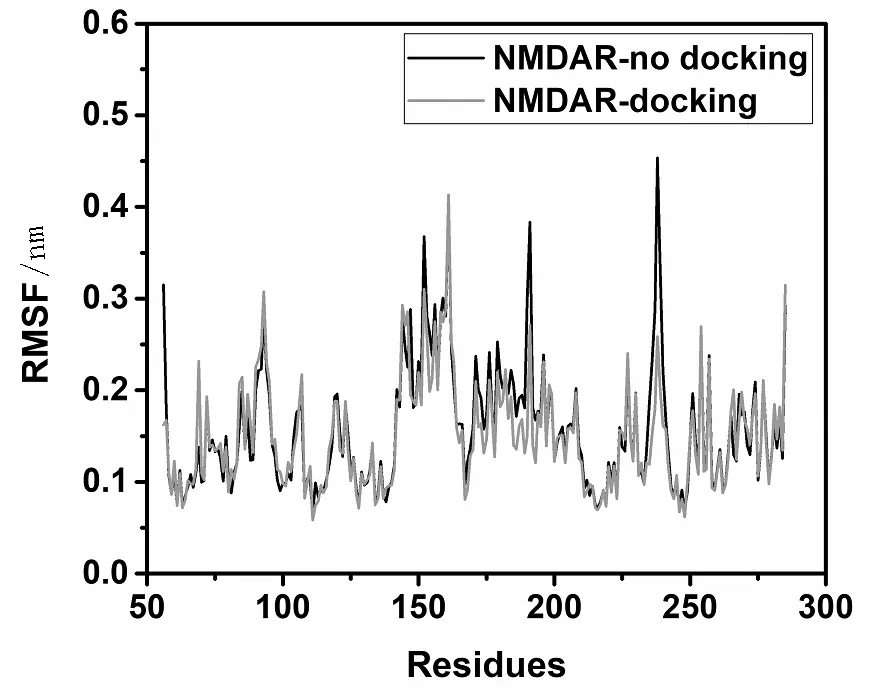
图3 NMDAR与艾司氯胺酮对接前后结构分子模拟过程中的RMSF图Fig.3 RMSF of NMDAR before and after docking with s-ketamine during molecular simulation
蛋白残基的均方根波动值(RMSF)是衡量残基在整个运动轨迹中相对于平均位置的波动程度。为了考察艾司氯胺酮的结合对NMDAR功能区域的影响,对NMDAR在结合前后分子模拟过程中各个残基的RMSF进行了分析,结果如图3所示。从图中可见,在艾司氯胺酮的结合之后,部分残基的RMSF值发生了明显的变化。其中,TRP238、THR56、GLY190、LYS239、LYS191、ARG237等残基的RMSF变化较大,表明艾司氯胺酮的结合可能主要影响与这些残基相关区域的功能,从而调节NMDAR的活性。
通过考察蛋白的溶剂可及表面积可以考察蛋白的亲水和疏水性。考察了NMDAR在与艾司氯胺酮对接前后的结构在分子模拟过程中溶剂可及表面积的变化,结果如图4所示。从图中可见,蛋白的溶剂可及表面积可分为亲水性表面积和疏水性表面积,NMDAR在与艾司氯胺酮对接之后,总的溶剂可及表面积有所减小,其中,疏水性表面积几乎无变化,而亲水性表面积减小,表明NMDAR在与艾司氯胺酮对接之后,蛋白的亲水性减小,这可能是艾司氯胺酮能够抑制NMDAR活性的原因之一。

图4 NMDAR与艾司氯胺酮对接前后分子模拟的溶剂可及表面积Fig.4 Solvent accessible surface of NMDAR before and after docking with s-ketamine during molecular simulation

图5 NMDAR与艾司氯胺酮对接前后分子模拟过程中蛋白内部的氢键数目Fig.5 Hydrogen bonds number inside NMDAR before and after docking with s-ketamine during molecular simulation
蛋白内部的氢键是影响蛋白稳定性的主要原因之一。为了考察艾司氯胺酮对NMDAR稳定性影响的原因,分析了MBDAR在与艾司氯胺酮对接前后蛋白内部氢键数目的变化,结果如图5所示。从图中可见,在与艾司氯胺酮对接之前,MBDAR内部的氢键数目在190个左右,在分子模拟的过程中变化不大,维持在170个至210个之间。在与艾司氯胺酮对接之后,MBDAR内部的氢键数目有所增加,并且在模拟的过程较为稳定,维持在180个至215个之间,表明MBDAR在与艾司氯胺酮对接之后蛋白内部氢键数目的增加可能是蛋白稳定的原因。
3 结论
采用分子对接和分子动力学方法研究了艾司氯胺酮与NMDAR之间的分子作用机制,结果表明,艾司氯胺酮趋向结合于NMDAR的口袋部位,使蛋白N端的运动方式从有序的顺时针运动改变为无序的运动,并使运动振幅减弱。经过分子模拟获取了NMDAR与艾司氯胺酮结合的稳定结构,发现艾司氯胺酮主要影响蛋白的TRP238、THR56、GLY190等残基部位。通过蛋白的溶剂可及表面积和蛋白内部氢键数目分析表明,艾司氯胺酮的结合使NMDAR的亲水性减小,疏水性增加,蛋白内部氢键数目增加,这可能是艾司氯胺酮调节NMDAR结构活性的主要因素,本文的研究为阐明艾司氯胺酮的抗抑郁机制和新型抗抑郁药物的开发提供了理论依据。
