IFT140 基因纯合突变致Mainzer-Saldino 综合征1 例报告并文献复习
2020-06-06魏海霞孙良忠林宏容岳智慧陈华木
魏海霞 孙良忠 林宏容 岳智慧 李 敏 陈华木
1.南方医科大学南方医院儿科(广东广州 510515);2.中山大学附属第一医院儿科(广东广州 510080)
Mainzer-Saldino 综合征(Mainzer-Saldino syndrome,MSS)是一种罕见的伴骨骼发育异常的肾单位肾痨相关纤毛病(nephronophthisis related ciliopathy,NPHP-RC),1970 年首次报道[1],又名Conerenal综合征,是一种常染色体隐性遗传性疾病,也是短肋胸廓发育不良(short rib-thoracic dysplasia,SRTD)综合征的一种类型[1-2]。MSS有严重视网膜营养不良、慢性肾衰竭、指(趾)骨圆锥状骨骼等症状,部分患者伴有头颅畸形、小脑共济失调、肝脏损害和生长发育迟缓等表现[1-3]。MSS 致病基因主要是编码初级纤毛内转运(intraflagellar transport,IFT)蛋白的基因,有IFT140和IFT172,临床最常见的是IFT140。IFT 140基因突变患者的临床表型较为复杂,从非综合征型视网膜病变到多系统损害的纤毛病,如MSS、Jeune 综合征、Sensenbrenner 综合征、Opitz 三角头综合征等[4-5],其中MSS 最常见。本文回顾性分析1 例IFT140纯合突变MSS患儿的临床特点及其基因变异特点,以提高临床医师对MSS的认识。
1 临床资料
患儿,女。3 岁时发现视力弱于常人;5 岁8 月龄因面色苍白10 月余,眼睑、双下肢水肿伴咳嗽6 天就诊。患儿系G2P2,足月顺产,出生时无产伤和窒息史。父母体健,非近亲结婚。哥哥生长发育正常。既往史及家族史无特殊。体格检查:身高110 cm(-0.8 SD),体质量19 kg,血压130/90 mmHg。重度贫血貌,头颅、胸廓无明显畸形,心肺腹无异常。手指粗短,末梢稍膨隆,呈杵状(图1)。辅助检查:血红蛋白41 g/L;血肌酐698.5 μmol/L,估算肾小球滤过率(eGFR)6.5 mL/(min·1.73 m2),尿素氮35.9 mmol/L,二氧化碳12.8 mmol/L;丙氨酸氨基转移酶13 U/L,天冬氨酸氨基转移酶26 U/L;钾5.58 mmol/L,钙1.34 mmol/L,磷2.28 mmol/L;血气分析 pH值 7.19,二氧化碳分压18.3 mmHg,碱剩余-20.1 mmol/L;尿比重1.005,尿蛋白++,尿红细胞阴性,尿白蛋白1 070.0 mg/L,尿β2微球蛋白40.9 mg/L,尿α1微球蛋白94.30 mg/L,尿蛋白/肌酐比值 4.29 mg/mg;心肌酶谱及凝血功能无异常。B超示双肾大小形态正常,左肾72 mm×24 mm,右肾78 mm×25 mm,双肾弥漫性病变,未见异常回声;肝胆胰脾无明显异常。眼底检查示双侧视网膜退行性病变(图1)。左腕骨正位片示骨化中心7个,骨龄6 岁,双手指中远节指骨短小,密度不均,部分硬化,各指骨多个骨骼呈锥状改变(图1)。
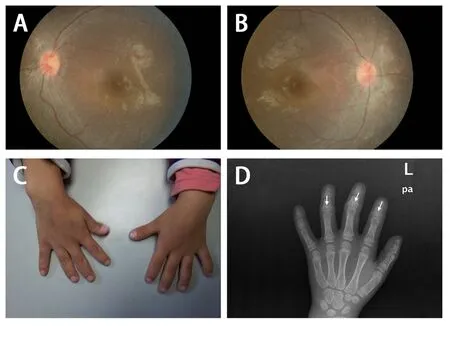
图1 患儿眼底和手指骨改变
综合患儿临床资料,疑诊为MSS。征得其父母知情同意及医院伦理委员会批准,收集患儿及其父母、哥哥外周静脉血各2 mL,分别提取基因组DNA送深圳华大基因股份有限公司进行全外显子测序(WES)。采用Agilent SureSelect Human All Exon51(V4)芯片进行捕获,Hiseq4000 平台进行上机测序。通过BWA与UCSChg19数据库进行比对。结果发现,患儿16号染色体上存在IFI140纯合突变c.634G>A,p.G212R。使用Polyphen2、Mutation taster、SIFT预测IFT140候选变异致病性、蛋白质结构功能变异预测均提示为致病性突变。Sanger 测序验证示突变分别来自患儿父母(图2)。此外,还检测到患儿携带1 个IFT 172单杂合突变c.1513 C >T,p.R 505 W。患儿哥哥未发现IFT140基因变异。
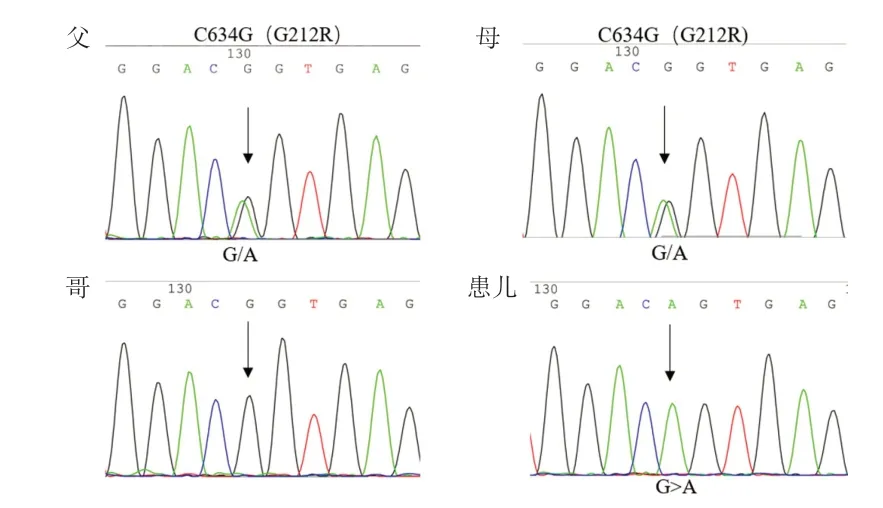
图2 患儿及家系IFT140 基因突变 Sanger 测序验证图
2 讨论
以“Mainzer-Saldino 综合征”为关键词或主题词检索万方、维普、中国知网数据库;以“Mainzer-Saldino syndrome”或“Conerenal syndrome”检索PubMed 和EBSCO数据库,检索时间1970至2020年1月。排除综述、动物实验文献,检索到中文文献1篇,英文文献35篇。共报道42个家系48例MSS患者,国内报道1例,集中在欧美地区,其中IFT140突变25例,IFT172突变4例,部分患者未行基因检测[6-9]。
MSS 较为罕见,目前报道的最小发病年龄为2岁。几乎所有MSS 患者都具有视网膜营养不良、慢性肾衰竭和指骨骨骼锥形改变的临床特点,部分患者有轻度的胸廓和小头畸形、髋骨软骨内骨化,伴生长发育迟缓、身材矮小等。MSS 的肾脏表现多数为少年型肾单位肾痨,影像学检查可表现为双肾外形大小正常或轻度缩小,皮髓质分化不清,病理改变为肾小管间质损伤,多数患者在13 岁内进入终末期肾衰竭[7]。IFT 140基因是第一个被证实导致MSS 的致病基因,位于染色体16p13.3,由31个外显子组成,编码蛋白IFT 140,是IFT-A(负责纤毛逆行运输)的核心成分,含有1 462个氨基酸,尾端含有重复色氨酸-天冬氨酸二肽(with a repeat trytophane-aspartic acid dipeptide,缩写WD)结构域(集中在蛋白质N 端)和9 个四肽重复序列(tetratricopeptide repeat,TPR)结构域(集中在蛋白质C端)。WD结构域主要参与纤毛的形成、信号转导及膜相关蛋白的组分,TPR结构域则在蛋白复合物的内部相互作用和稳定中具有重要作用[10]。IFT140功能障碍将导致纤毛变短,纤毛逆行运输受阻,从而导致多种纤毛病功能障碍。有报道11 例外显子15 的纯合突变c.1990G>A(p.Glu664Lys)患者,来自近亲结婚家系,均表现为非综合型视网膜病变[11],但也有报道该位点纯合突变的患者表现为MSS[11-12]。同一突变位点可有不同表现,推测可能与表观遗传因素或其他的基因相互作用有关。也有学者认为,IFT140突变位点与临床表型可能存在一定的相关性:MSS、Jeune 综合征患者突变位点多数位于WD 结构域;非WD 结构域或TPR结构域突变对应的临床表型较轻,如非综合征型视网膜病变[4]。目前国内已报道中国人群IFT 140基因突变8 例,均为复合杂合突变,3 例患者表现为非综合型视网膜病变,另3 例则为先天性黑曚症,以上6 例患者均为非综合征型视网膜病变,仅表现为眼部损害,无其他器官受累[4,12]。1 例9 岁患儿则表现为MSS,另1 例27 岁的男性患者临床表现仅为不育,无其他系统受累表现,提示IFT140基因的作用较为广泛,基因型与表型的关系仍待进一步挖掘[13-14]。
本例患儿3 岁时以视力异常为首发症状,到5 岁就诊时已进入终末期肾病。眼底检查示视网膜退行性病变,X 线片呈指骨骨骼锥状改变,肾脏B 超示双肾弥漫性病变,符合典型的MSS临床表现。基因检测发现突变位点位于16 号染色体上IFT 140基因外显子6的c.634G>A纯合突变导致氨基酸改变p.G212R,为文献已报道的致病性突变[8,15]。综合临床表现和基因检测结果,MSS 诊断成立。患儿父母均为携带者,哥哥未发现该变异,父母及哥哥均无临床表现,符合常染色体隐性遗传的发病规律。曾有报道1例与本例患儿完全一致的IFT140纯合突变,临床表型也是MSS,但其突变来源于罕见的节段性单亲二倍体(母方)[15]。另有报道包含该位点变异的复合杂合突变患者表现为Jeune综合征[8]。与MSS相比较,Jeune综合征具有严重的致死性胸廓发育不良,导致严重的呼吸窘迫。本例患儿胸廓明显畸形改变,故可鉴别。
综上所述,特征性的指趾骨改变、先天性视网膜发育不良和进行性肾功能损害对诊断MSS 有提示作用,对疑似患者及时进行基因检测有助于临床早期明确诊断、指导治疗和遗传咨询。
