HPLC测定甲硝唑葡萄糖注射液中甲硝唑含量分析方法验证
2020-06-05李晓路
文/李晓路
对甲硝唑葡萄糖注射液中甲硝唑含量的测定采用HPLC测定分析方法进行方法学验证 // 采用高效液相色谱法(HPLC)对甲硝唑葡萄糖注射液中甲硝唑含量进行测定,甲硝唑在150.24~350.56 µg/mL范围内线性关系良好,其r值为0.9995,平均回收率为99.98%,回收率RSD为0.22%(n=9)。通过对其专属性、回收率(准确度)、线性、精密度、稳定性等试验,证明该方法简便灵敏、准确可靠、重现性好,HPLC可作为甲硝唑葡萄糖注射液中甲硝唑含量测定方法。
甲硝唑为抗原虫及抗厌氧菌药,甲硝唑葡萄糖注射液广泛应用于临床,其甲硝唑含量在《中国药典》(2005年版二部)中采用紫外-可见分光光度法直接测定,不能排除葡萄糖分解产物5-羟甲基糠醛的干扰。《中国药典》(2010年版二部)规定甲硝唑葡萄糖注射液中甲硝唑含量采用高效液相色谱法进行检测,能有效的排除5-羟甲基糠醛的干扰。通过对甲硝唑含量测定所用的HPLC分析方法进行方法学验证,以证明该方法简便灵敏、准确可靠、重现性好,高效液相色谱法(HPLC)可作为甲硝唑葡萄糖注射液中甲硝唑含量测定方法。
实验部分
仪器与试药
1.仪器及色谱条件
岛津LC-20A高效液相色谱仪(配备LC solution工作站)、岛津SPD-20A紫外检测器;
色谱柱:十八烷基硅烷键合硅胶为填充剂(VP-ODS 250*4.6 mm);
流动相:甲醇-水(20:80);
检测波长:320 nm;
理论板数:按甲硝唑峰计算不低于2000。
2.试药
甲硝唑对照品;
甲硝唑葡萄糖注射液;
甲醇为色谱纯;
实验用水为纯化水;
其他试剂均为分析纯。
实验过程
1.专属性
专属性是指在样品介质中有其它组份(如杂质、降解产物、辅料)共存时,采用的检验方法(如鉴别检查、杂质检查、含量测定)能准确测定出被测物质的特性。
空白溶液:按处方配制不含甲硝唑的溶液,作为空白溶液。取空白溶液及供试品溶液,照含量测定项下方法分别测定,检测结果见表1,谱图见图1。空白溶液对含量测定应无干扰。
空白溶液制备:按处方配制不含甲硝唑的溶液,精密量取该溶液25 mL,置200 mL量瓶中,用流动相稀释至刻度,摇匀即得。
供试品溶液制备:精密量取甲硝唑葡萄糖注射液25 mL,置200 mL量瓶中,用流动相溶解并稀释至刻度,摇匀即得,照含量测定项下方法分别测定,检测结果见表2,谱图见图2。
通过上述数据显示:空白溶液对含量测定无干扰。
2.准确度
准确度系指该方法测定的结果与真实值或参考值接近的程度,一般用回收率表示。一般要求分别配制浓度为80%、100%和120%的供试品溶液各三份,分别测定其含量,将实测值与理论值比较,计算回收率。平均回收率应在98.0%~102.0%之间,相对标准偏差(RSD)应不大于2.0%。
对照品溶液的制备:称取经105℃干燥2小时的甲硝唑对照品25mg,精密称定,置100 mL量瓶中,加流动相溶解并稀释至刻度,摇匀即得 。
供试品溶液制备:称取经105℃干燥2小时的甲硝唑对照品约20 mg、25 mg、30 mg,精密称定,分别置100 mL量瓶中,用流动相溶解并稀释至刻度,即得浓度为80%、100%和120%的供试溶液,各浓度各配置3份,共9个样品,结果见表3。
计算公式:

式中:
A样—供试品的峰面积或峰高;
A对—对照品的峰面积或峰高;
W—对照品的重量(mg)。
通过上述数据显示:平均回收率在98.0%~102.0%之间,相对标准偏差(RSD)小于2.0%。故该方法有较好的回收率,具有良好的准确度。
3.线性
线性是指在设定范围内,测试结果与试样中被测物质浓度直接成正比关系的程度。线性一般通过线性回归方程的形式来表示。具体的验证方法为:在60%至140%的浓度范围内配制至少5份浓度不同的对照品溶液,分别测定其主峰的面积,计算相应的浓度。以浓度C为横坐标(X轴),峰面积A为纵坐标(Y轴)进行线性回归,计算回归方程,得出相关系数r值。标准曲线的相关系数(r)不得小于0.9990。
样品溶液制备:精密称取甲硝唑对照品25.04 mg置50 mL量瓶中,加流动相溶解并稀释至刻度,摇匀,量取该样品3 mL、4 mL、5 mL、6 mL、7 mL,分别置10 mL量瓶中,加流动相稀释并定容至刻度,摇匀,即分别得浓度为60%、80%、100%、120%和140%的 供试品溶液,检测结果见表4。

表1 专属性空白溶液峰表

图1 专属性空白溶液谱图
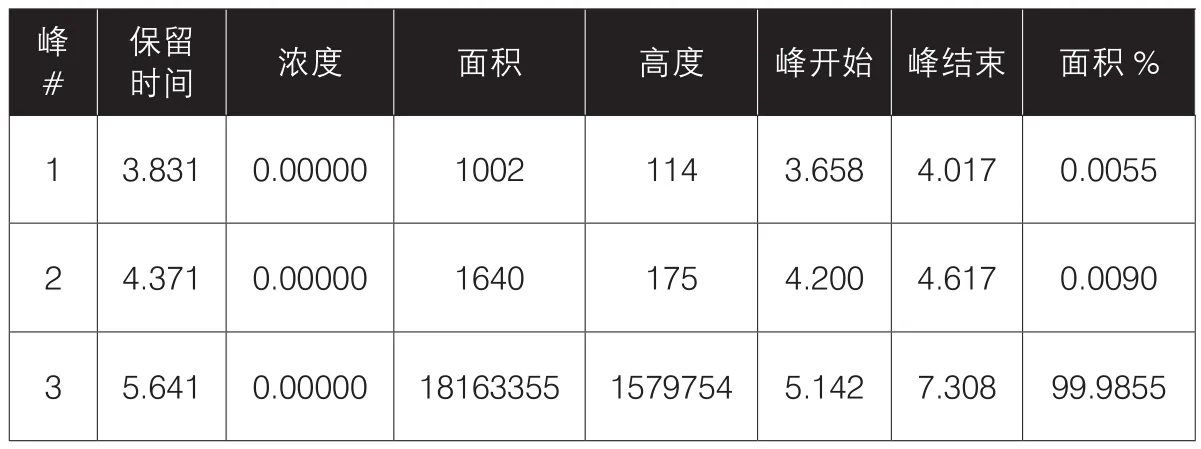
表2 专属性供试品溶液峰表

图2 专属性供试品溶液谱图
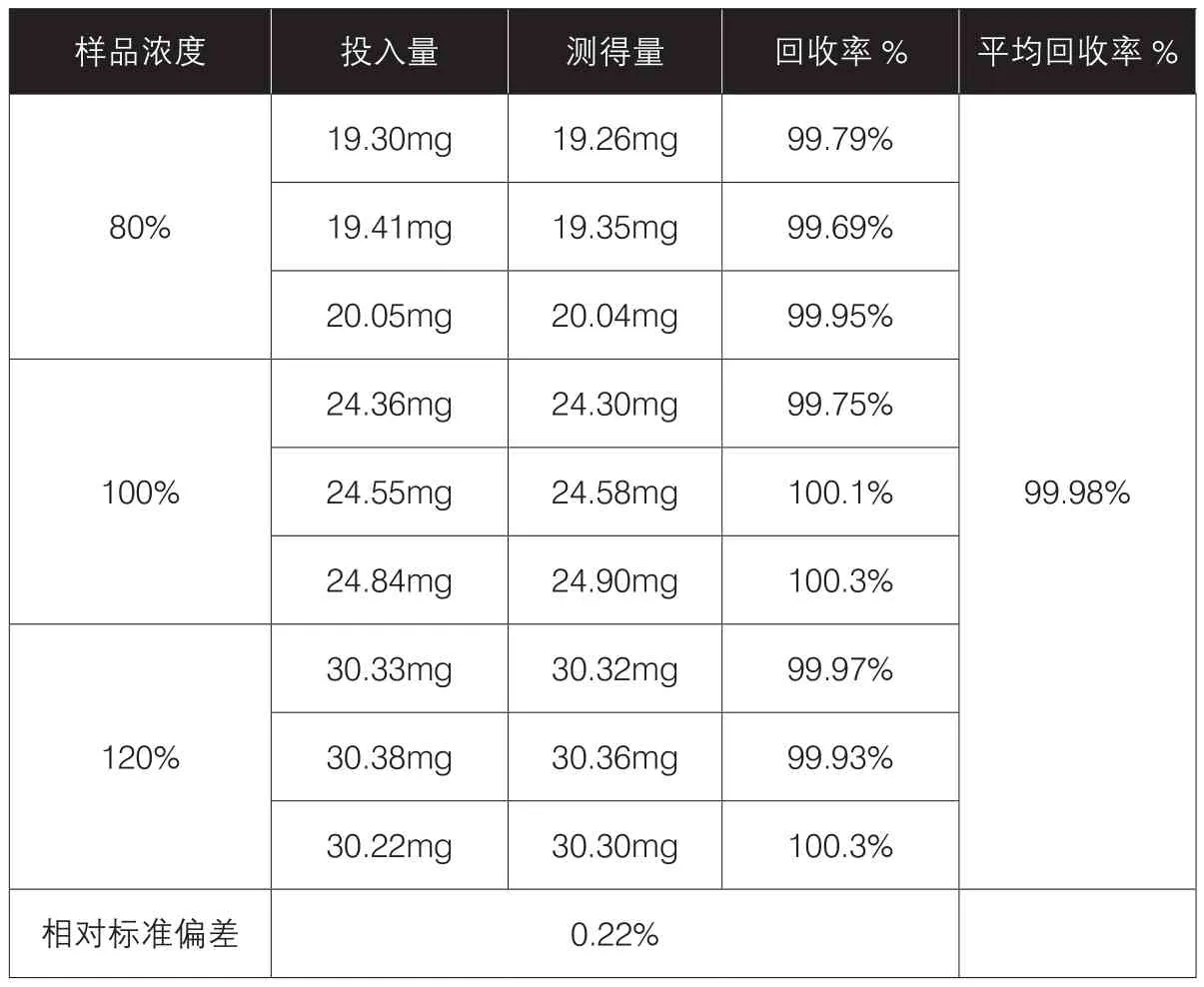
表3 80%、100%和120%三种浓度供试品溶液的含量、回收率及相对标准偏差

表4 5份浓度不同的对照品溶液的主峰面积
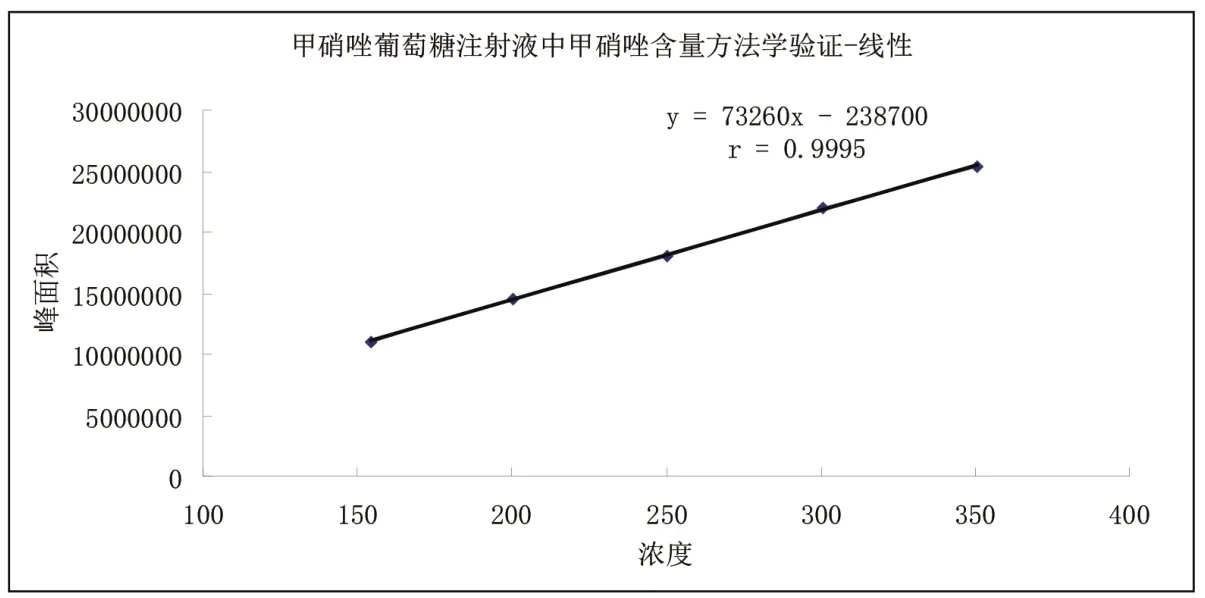
图3 线性关系标准曲线图
计算公式:

式中:
W—对照品的重量(mg);
V—量取样品的体积(mL)。
通过线性回归方程计算r=0.9995,甲硝唑溶液在154.24~350.56 µg/mL 范围内呈良好的线性关系,见图3。
4.精密度
精密度系指在规定的测试条件下,同一个均匀供试品,经多次取样测定所得结果之间的接近程度。一般用偏差、标准偏差或相对标准偏差表示。
■重复性
照含量测定项下方法,由同一个分析人员在尽可能相同的条件下连续测定6次,所得6组含量结果的相对标准偏差应小于2.0%。
对照品溶液的制备:精密称取经105℃干燥2小时的甲硝唑对照品13.03 mg,置50 mL量瓶中,加流动相溶解并稀释至刻度,摇匀即得。
供试品溶液制备:精密量取甲硝唑葡萄糖注射液25 mL,置200 mL量瓶中,用流动相溶解并稀释至刻度,摇匀即得,结果见表5。
计算公式:

式中:
A样—供试品的峰面积或峰高;
A对—对照品的峰面积或峰高;
W—对照品的重量(mg);
2—每mL含甲硝唑的量(mg)。
通过表中数据得知:同一分析人员所得6组含量结果的相对标准偏差小于2.0%,该方法有较好的重复性。
■中间精密度
照含量测定项下方法,由不同分析人员在不同时间分别测定含量,至少测定6次,用测得的数据计算,其相对标准偏差应小于2.0%。
对照品溶液的制备:精密称取经105℃干燥2小时的甲硝唑对照品13.02 mg,置50 mL量瓶中,加流动相溶解并稀释至刻度,摇匀即得 。
供试品溶液制备:精密量取甲硝唑葡萄糖注射液25 mL,置200 mL量瓶中,用流动相溶解并稀释至刻度,摇匀即得,结果见表6。
计算公式:

式中:
A样为供试品的峰面积或峰高;
A对为对照品的峰面积或峰高;
W为对照品的重量(mg);
2为每ml含甲硝唑的量(mg)。
通过表中数据得知:不同分析人员所得12组含量结果的相对标准偏差小于2.0%,该方法有较好的中间精密度。
5.稳定性试验
按含量测定项下供试品配制方法配制供试品溶液,分别于0、2、4、6、8小时测定供试品溶液,记录色谱图,以峰面积计算相对标准偏差(RSD),结果见表7。
接受标准:稳定性试验相对标准偏差(RSD)应小于2.0%。
通过表中数据所得:稳定性试验相对标准偏差(RSD)小于2.0%,证明供试品溶液在8小时内稳定性良好。
6.耐用性
耐用性是指在测定条件有小的变动时,测定结果不受影响的承受程度,为使方法可用于常规检验提供依据。分别考察流动相比例变化±5%、柱温变化±5℃,仪器色谱行为的变化,每个条件下各测试两次。
可接受的标准为:主峰的拖尾因子不得大于2.0,主峰与杂质峰必须达到基线分离;各条件下的含量数据(n=4)的相对标准偏差应不大于2.0%。
对照品溶液的制备:精密称取经105℃干燥2小时的甲硝唑对照品13.09 mg,置50 mL量瓶中,加流动相溶解并稀释至刻度,摇匀即得 。
供试品溶液制备:精密量取甲硝唑葡萄糖注射液25 mL,置200 mL量瓶中,用流动相溶解并稀释至刻度,摇匀即得,结果见表8。
在流动相比例变换±5%、柱温变化±5℃,主峰的拖尾因子不大于2.0,主峰与杂质峰达到基线分离;各条件下的含量数据(n=4)的相对标准偏差应小于2.0%,故该方法有较好的耐用性。
7.定量限
称取甲硝唑对照品约10 mg,精密称定,用流动相反复稀释,按上述色谱条件进样,根据定量限的信噪比为10:1的原则,最终确定甲硝唑的定量限。

表5 供试品溶液的重复检测结果

表6 供试品中间浓度的重复检测结果

表7 稳定性试验
供试品溶液的制备:1号溶液:精密称取甲硝唑对照品13.07 mg,置100 mL量瓶中,加流动相溶解并稀释至刻度,摇匀。
2号溶液:精密量取1号溶液1 mL,置100 mL量瓶中,加流动相稀释至刻度,摇匀,滤过,精密量取续滤液10 µL,注入液相色谱仪,记录色谱图。
3号溶液:精密量取2号溶液5 mL,置50 mL量瓶中,加流动相稀释至刻度,摇匀,滤过,精密量取续滤液10 µL,注入液相色谱仪,记录色谱图。
4号溶液:精密量取3号溶液5 mL,置50 mL量瓶中,加流动相稀释至刻度,摇匀,滤过,精密量取续滤液10 µL,注入液相色谱仪,记录色谱图。

由上述数据及图谱可知:该方法有较好的定量限。
8.检测限
精密量取定量限项下供试品溶液3 mL,置10 mL量瓶中,用流动相稀释至刻度,摇匀,按上述色谱条件连续进样,根据检测限的信噪比为3:1的原则,确定甲硝唑的检测限与最小检测浓度。
供试品溶液的制备:精密量取定量限项下的供试品溶液3 mL置10 mL量瓶中,用流动相稀释至刻度,摇匀,滤过,精密量取续滤液10µL,注入液相色谱仪,记录色谱图。供试品溶液色谱图中信噪比约为3:1,可确定甲硝唑的检测限与最小检测浓度。

由上述数据及图谱可知:该方法有较好的检测限。
9.系统适用性
照含量测定项下方法配制对照品溶液1份, 连续进样5次,主峰峰面积的相对标准偏差应不大于2.0%,主峰保留时间的相对标准差应不大于1.0%。另外,主峰的拖尾因子不得大于2.0,主峰与杂质峰必须达到基线分离,主峰的理论塔板数应符合质量标准的规定。
对照品溶液的制备:精密称取经105℃干燥2小时的甲硝唑对照品13.06 mg,置50 mL量瓶中,加流动相溶解并稀释至刻度,摇匀即得,检测结果见表9。
通过表中数据所得:主峰峰面积的相对标准偏差小于2.0%,主峰保留时间的相对标准差小于1.0%,主峰的拖尾因子小于2.0,主峰与杂质峰达到基线分离,主峰的理论塔板数符合质量标准的规定。

表8 连续进样5次的相对标准偏差
10.含量测定
精密量取本品25 mL,置200mL量瓶中,用流动相定量稀释制成每1 mL中约含甲硝唑0.25 mg的溶液,摇匀,精密量取10 µL,注入液相色谱仪,记录色谱图;另取甲硝唑对照品适量,精密称定,加流动相溶解并稀释制成每1 mL中约含甲硝唑0.25 mg的溶液,摇匀,同法测定。按外标法以峰面积计算,即得。按上述色谱条件进行仪器设备调试,同法作平行试验,检测结果见表10。
11.结果计算
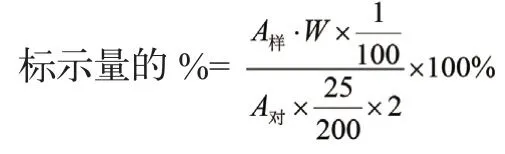
式中:
A样供—试品的峰面积或峰高;
A对—对照品的峰面积或峰高;
W—对照品的重量(mg);
2—每ml含甲硝唑的量(mg)。
由上述结果数据可知,两份平行试验结果相对偏差不得大于1.5%,该含量检测方法稳定可靠。
结论
甲硝唑葡萄糖注射液中甲硝唑含量采用高效液相色谱法进行检测,能有效的排除5-羟甲基糠醛的干扰。通过对分析方法专属性、回收率(准确度)、线性、精密度(重复性和中间精密度)、稳定性、耐用性、系统适用性、检测限、定量限等试验证明高效液相色谱法对甲硝唑葡萄糖注射液中甲硝唑含量的测定有足够的专属性,该方法简便灵敏、准确可靠、重现性好,可作为甲硝唑葡萄糖注射液中甲硝唑含量测定方法。

表9 连续进样5次的相对标准偏差

表10 双样双平行的含量测定
