Al掺杂Mg2Ge光电性质的第一性原理计算
2020-05-13姚秋原周卢玉侯亮亮
姚秋原, 谢 泉, 周卢玉, 余 宏, 侯亮亮
(贵州大学 大数据与信息工程学院 新型光电子材料与技术研究所, 贵阳 550025)
1引 言
近年来Mg2Ge因其在光电器件、发光器件、热电器件中的潜在应用前景而引起人们的关注,Mg2Ge具有高热稳定性、低密度、优良的压缩性、丰富的可用性、无毒性、低成本等优点[1-6].而掺杂是一种重要的调控能隙和光电性质的手段,为了使Mg2Ge在光电和热电领域有更广的应用,对Mg2Ge的掺杂研究成为近年来备受瞩目的研究热点.例如刘慧英[7]等人在对Mg2Ge作为锂离子电池电极材料研究中发现,随着Li嵌入量的增加,主体材料发生了从半导体性到金属性、又到半金属性的转化;李开跃[8]等人通过第一性原理计算和半经典玻尔兹曼理论研究了Mg2GexSn1-x(x=0.25,0.5,0.75)固溶体的电子结构和热电性质,发现n型Mg2GexSn1-x固溶体有望成为中温热电材料;ESAKA等人[9]运用气相沉积法在铜箔衬底上制备了不同厚度的Mg2Ge阳极,结果表明其对高能量和高功率密度的二次电池具有重要的应用价值.但是对于不同浓度Al掺杂Mg2Ge光电性质的研究报道极少,所以本文运用第一性原理计算了Al掺杂Mg2Ge的光电性质,为在Mg2Ge的掺杂方面提供了理论和实验的参考.
2计算方法
Mg2Ge是具有反萤石结构的化合物半导体,空间点群为Fm3m(No.255),晶格常数为0.638 nm,其中Ge4+离子和Mg2-离子分别位于萤石结构中立方原始单胞的(0,0,0)位置和±(1/4,1/4,1/4)位置,且Ge4+离子的配位数是8,Mg2-离子在Ge4+离子内部形成立方体结构.其晶体结构如图1所示.计算时,选取Mg2Ge原胞为主体,分别建立1×4×1、1×2×1、1×1×1超胞,用Al原子取代其中的一个Mg原子,分别得到Mg2-xAlxGe(x=0.125),Mg2-xAlxGe(x=0.25)及Mg2-xAlxGe(x=0.5)晶体结构.
文中计算所用到的是CASTEP软件包(Cambridge Serial Total Energy Package in Material Modeling Accelrys),利用基于密度泛函的第一性原理进行相关计算,是当前在电子结构计算中较为准确的理论方法.首先采用BFGS(Broyden-Fletcher-Goldfarb-Shanno)算法对晶体模型进行结构优化,能量截断值(energy cutoff)为380 eV,迭代收敛精度(SCF)为5×10-7eV/atom,离子实和价电子间的互相作用用超软赝势(ultrasoft)处理,关联泛函用广义梯度近似(GGA)处理,体系电子波函数通过平面波基组展开;在总能量的计算中,对于1×4×1、1×2×1、1×1×1晶体结构布里渊区积分分别采用了7×2×7、7×3×7、7×7×7的Monkhorst-Pack形式的高对称特殊k点方法,能量计算都在倒易空间中进行[10,11].
图1 Mg2Ge的晶体结构示意图Fig. 1 Schematic diagram of the crystal structure of Mg2Ge
3结果与讨论
3.1晶胞的几何优化
表1为进行几何结构优化后的Mg2-xAlxGe的晶格常数、晶胞体积与体系能量.未掺杂的晶格常数与实验值6.393 nm[12]相差不大,说明参数设置合理.从表中可以看出随着Al的浓度增加,晶胞体积逐渐减小,这是因为Al的原子半径小于Mg的原子半径,而Al在Mg2Ge体系中属于替位式杂质,取代了Mg原子,所以随着掺入量的增大晶胞体积逐渐减小.从体系总能量可以看出,纯Mg2Ge的能量最小所以最稳定,而随着Al掺杂量的增大体系越来越不稳定,掺杂难度逐渐增大,说明较低浓度的Al掺杂Mg2Ge材料更容易制备.

表1 Mg2-xAlxGe的晶格常数、晶胞体积和总能量
3.2能带结构
图2显示了不同掺杂浓度下Mg2-xAlxGe的能带结构.图2(a)为x=0时Mg2-xAlxGe的费米面附近的能带结构,图中选取禁带中点作为费米能级.W、L、G、X、Κ为第一布里渊区的高度对称点,在k空间的坐标是:W(0.500, 0.250,0.750),L(0.500,0.500,0.500),G(0.000,0.000,0.000),X(0.500,0.000,0.500),Κ(0.500,0.250,0.750).Mg2Ge价带中能级的最高点与导带中能级的最低点不在相同的k点位置,而是价带最大值位于G点,其值为0 eV,导带在X点取得最小值,其值为0.2136 eV,因此Mg2Ge具有Gv—Xc带隙为0.2136 eV的间接带隙.图2(b),(c),(d)分别为掺杂x=0.125,x=0.25,x=0.5时的能带图,可以看出掺杂时引入了更密集的能级,费米面从禁带中线进入导带中,表示掺杂后的Mg2Ge为n型半导体.这是因为Al离子半径和Mg离子半径差不多,掺入的Al原子取代了原来位置的Mg原子,然后电离产生导电电子并成为施主杂质.
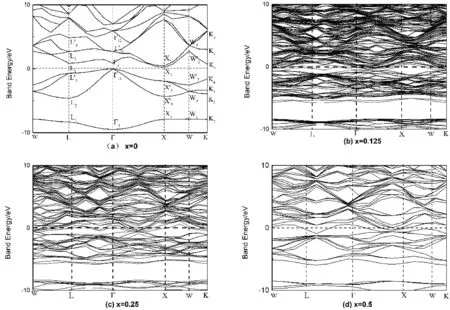
图2 不同掺杂浓度下的Mg2-xAlxGe能带结构Fig. 2 Mg2-xAlxGe band structure at different doping concentrations
3.3电子态密度
图3(a),(b),(c),(d)分别为x=0,x=0.125,x=0.25,x=0.5时Mg2-xAlxGe的总态密度(DOS)和各亚层电子的部分能态密度(PDOS).从图中可以看出,在未掺杂时Mg2Ge价带主要由Ge的4s、4p态电子和Mg的3s、3p态电子组成;费米能级附近的导带主要由Ge的4s态电子和Mg的3s、3p态电子组成,而Ge的4p态电子贡献较小.在掺杂后,Mg2-xAlxGe在费米能级附近的导带变成主要由Al的3p态电子、Ge的4s态电子和Mg的3s、3p态电子组成.并且掺杂后费米能级进入导带,材料变为n型导电.
3.4复介电函数
材料的带间跃迁微观物理过程可以通过介电函数与固体电子结构联系起来,固体的能带结构和各种光谱信息都可以从介电函数中得到映射.半导体材料Mg2Ge因为具有反萤石结构所以其光学性质是各向同性的,其介电函数是由不同能量的电子跃迁产生的,介电函数峰值的含义可以通过Mg2-xAlxGe的能带结构和态密度得到解释.
图4(a),(b)分别是Mg2-xAlxGe介电函数的实部ε1、虚部ε2与光子能量关系的图.从图4(a)中可以看出,随着掺杂量的增加Mg2-xAlxGe的静介电常数ε1(0)依次是:23.93,233,63.720,30.014,掺杂后ε1(0)均比未掺杂大,意味着掺杂后Mg2Ge的折射系数得到了较大提高,这和折射率图5(a)结果是一致的.从图4(b)中可以看出,与未掺杂相比掺杂后介电函数虚部的第一峰值均向低能方向偏移,这是因为Al作为施主杂质电离后释放大量电子,游离态的电子极易被极化.
3.5复折射率
复折射率是吸收性介质最主要的光学常数,由实部和虚部构成,实数部分n表示光波在吸收性介质中的传播速率,称为吸收性介质的折射率;虚数部分x叫做消光系数,表示光波在吸收性介质中传播时的造成的能量损失的多少.复折射率的实部,虚部和介电函数关系可由以下方程表示:
(1)
(2)
Mg2-xAlxGe复折射率的实部n和虚部x随光子能量变化的曲线图如图5(a),(b)所示,从图5(a)可以得出,纯Mg2Ge的折射率n0=4.5043,最大值在光子能量为2.7832 eV处取得,之后随着光子能量的增大折射率n开始逐步减小.Al掺杂后的n0均增大,其值随掺杂量的增加分别为:15.049,7.997,5.479,在x=0.125时取得极大值.由图5(b)可以看出,纯Mg2Ge的消光系数 最大值在光子能量为2.513 eV处取得,之后随着能量的增大而逐渐减小,掺入Al后消光系数 向低能方向偏移且最大值随掺杂浓度的增加呈现先增大后减小的趋势.
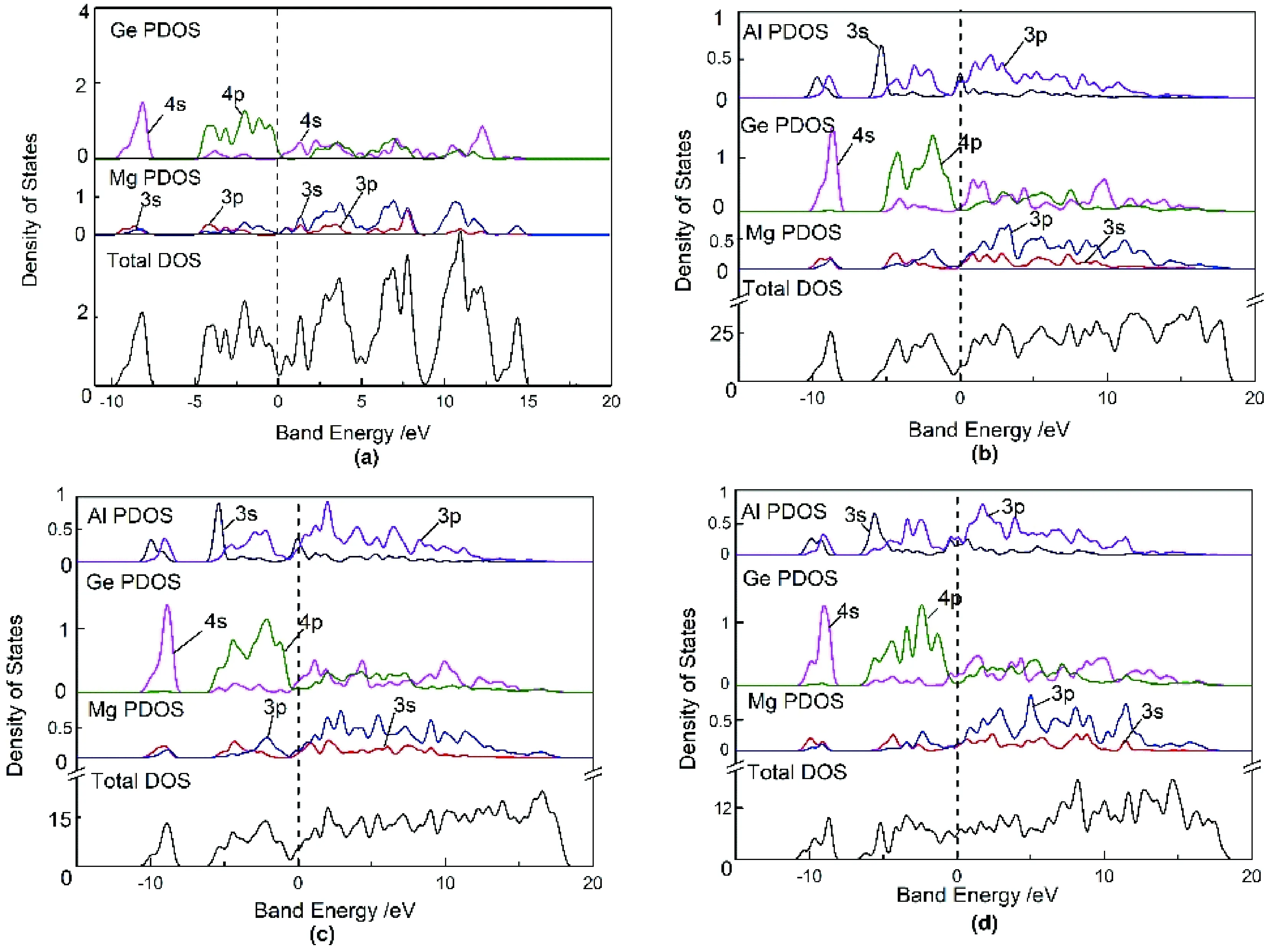
图3 不同掺杂浓度下的Mg2-xAlxGe态密度Fig.3 Mg2-xAlxGe state densities at different doping concentrations
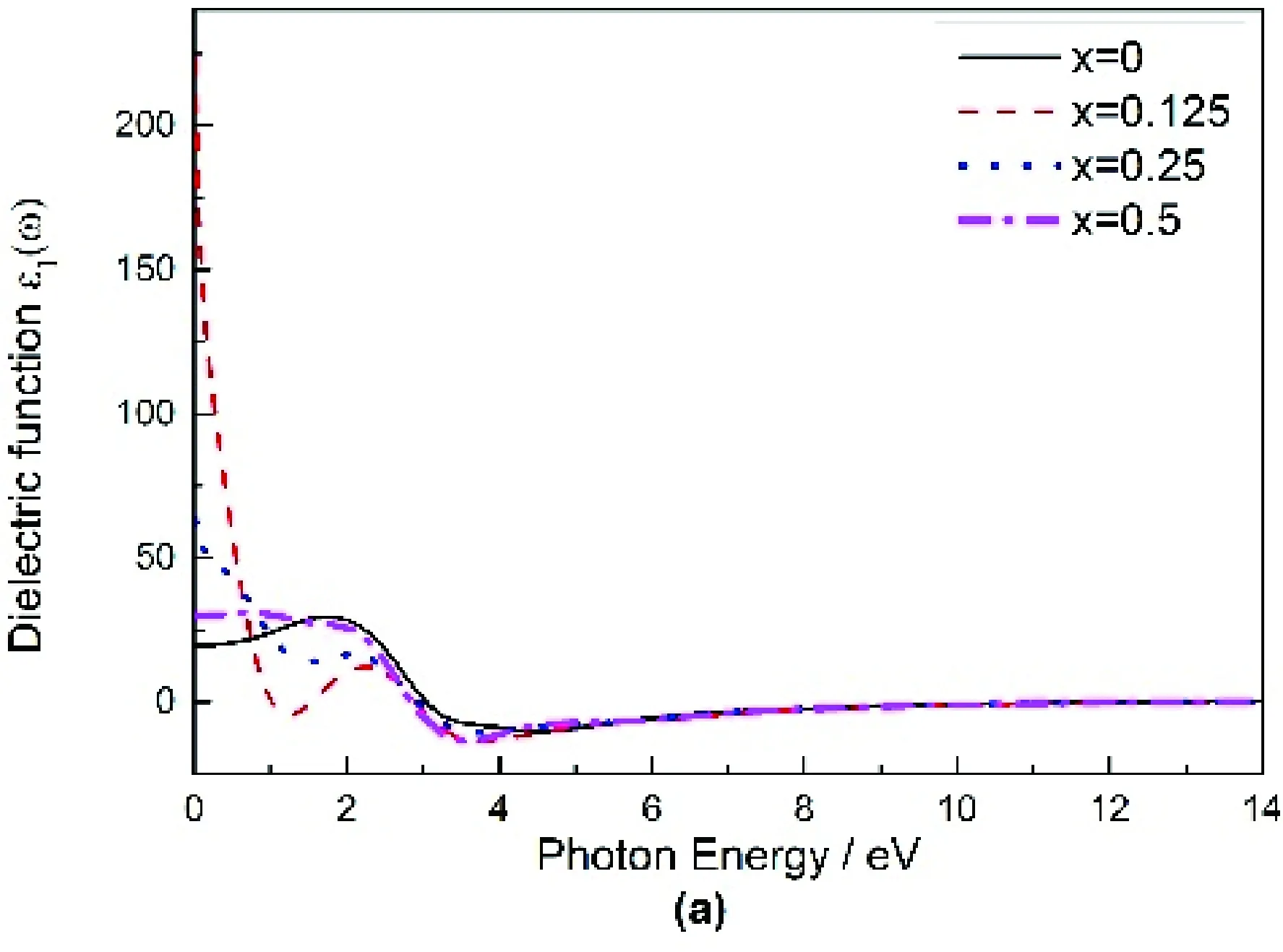

图4 Mg2-xAlxGe复介电函数的虚部和实部
3.6吸收系数

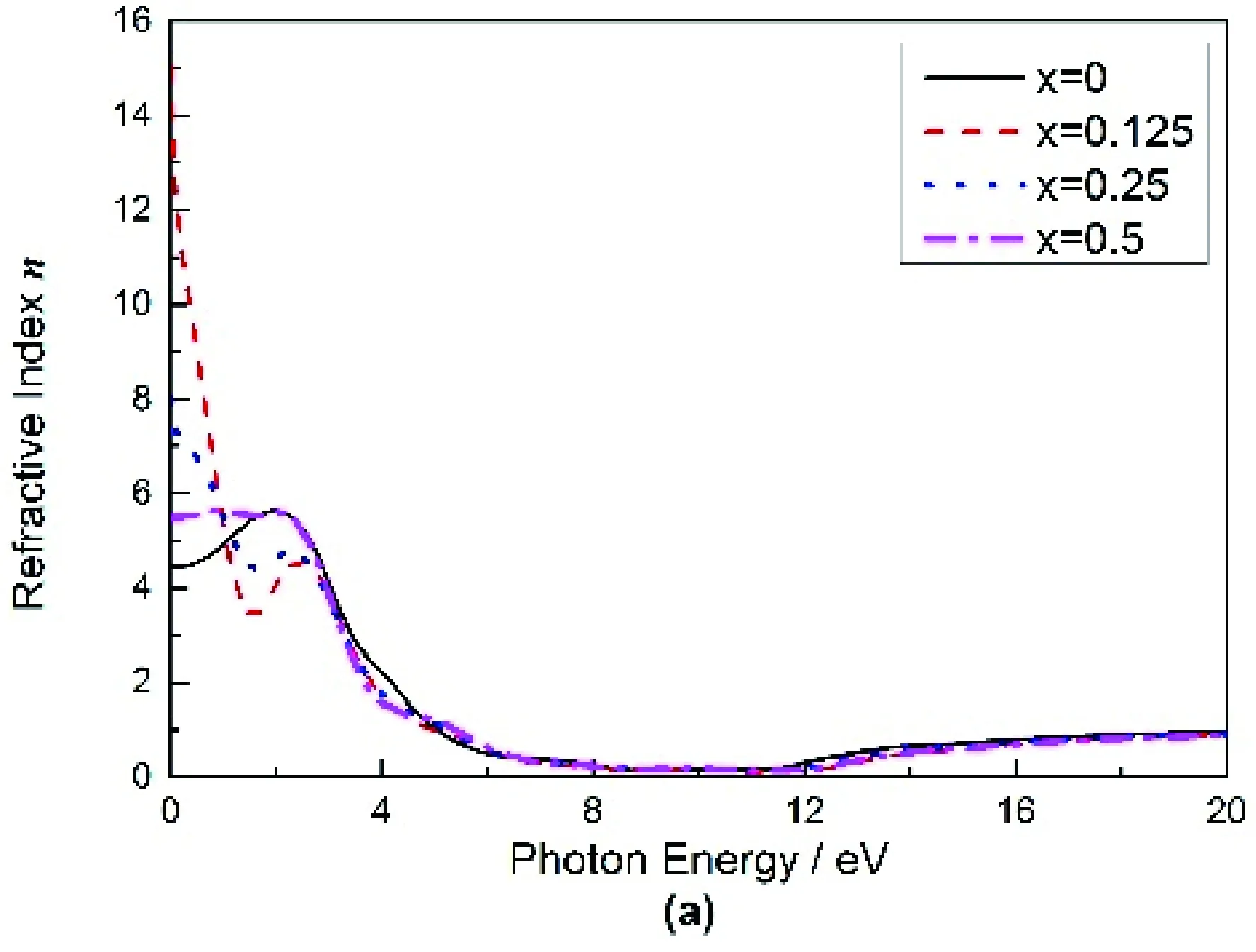
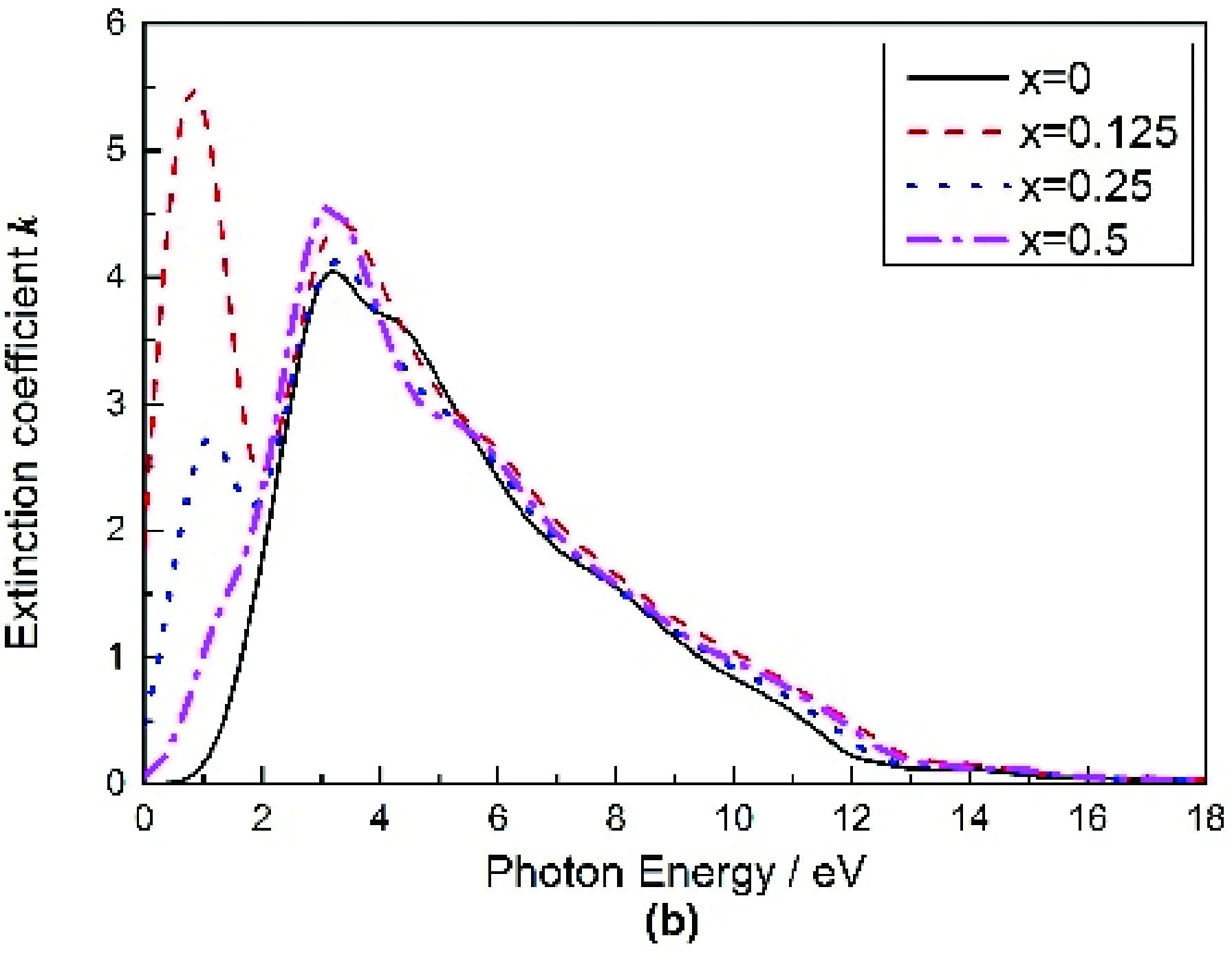
图5 Mg2-xAlxGe的折射率和消光系数Fig. 5 The refractive indexes n and extinction coefficients of Mg2-xAlxGe
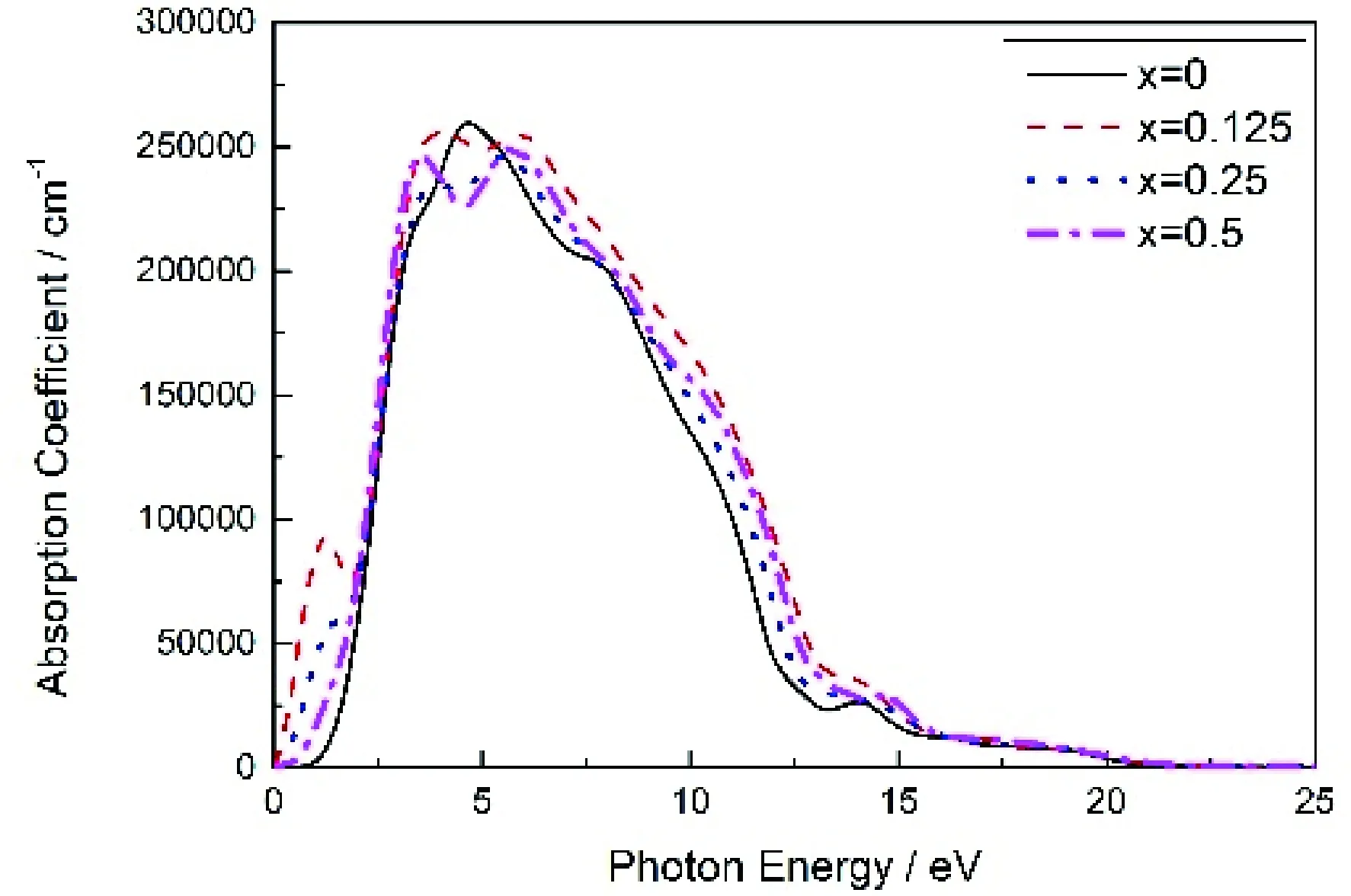
图6 Mg2-xAlxGe的吸收系数Fig. 6 The absorption coefficients of Mg2-xAlxGe
3.7反射系数
当具有复折射率的介质被光经由空气以入射角90度射到时,这时n1=1,n2=n+ik即得到反射率和复折射率的函数关系式:
(3)
Mg2Ge的反射率随光子能量变化的关系如图7所示,从图中可以得到纯Mg2Ge在能量为5 eV到11 eV的区间内很大一部分入射光都被反射,折射率n的值就相对很小,Mg2Ge呈现出金属反射特性.掺入Al后发现,在能量为02eV和1012 eV范围内,反射率得到明显提升,说明掺杂后的Mg2Ge金属反射特性增强,并且在x=0.125时反射系数变化率取得极大值.

图7 Mg2-xAlxGe的反射谱Fig. 7 The reflectance spectrums of Mg2-xAlxGe
3.8光电导率
当外界存在光照时,材料内部一部分低能级的电子会吸收能量而跃迁至导带中成为自由电子,导致电导率发生变化,这种由光注入引起的电导率发生变化的现象称为光电导效应.光电导效应广泛应用于现代科学技术的各个方面如辐射的探测与测量,太阳能电池中光电能量转换等.
Mg2-xAlxGe的复光电导率的实部1()随光子能量变化的曲线图如图8所示,与图4(b)中Mg2-xAlxGe的复介电函数虚部ε2相比较发现两者是相对应的.对于纯Mg2Ge在光子能量未达到0.8702 eV之前光电导率为0,之后随着能量的增加而逐步增大,在光子能量达到2.4280 eV时取得最大值,这时光电导率变化主要由带间激发跃迁导致,与Mg2Ge的能带和态密度对比得到参与带间跃迁的电子主要是Ge的4p态电子向Mg的3p态电子的跃迁.掺入Al后发现,除了x=0.125时光电导率峰值相对未掺杂时有所增大,其余掺杂浓度光电导率峰值均减小,而且在x=0.125时光电导率峰值位置向低能偏移,这是因为Al作为施主杂质在束缚态时周围有多余的价电子,而这些价电子只要很少能量就可以挣脱束缚成为导电电子,从而提高了载流子浓度和电导率;而掺杂浓度越大,晶格缺陷越多,非平衡载流子寿命越短,进而使电导率减小.
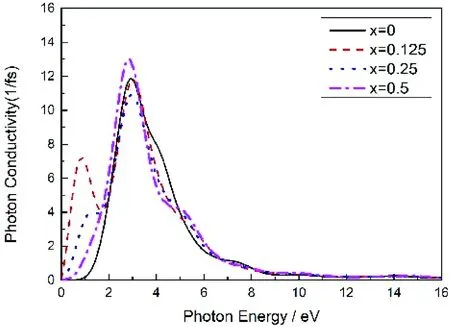
图8 Mg2-xAlxGe的光电导率Fig. 8 The complex photoconductivities of Mg2-xAlxGe
3.8能量损失函数
能量损失函数也与介电函数有特定的函数关系,关系式如下:
(4)
它可以表示均匀电介质对穿过其内部的电子能量吸收的情况.图9表示Mg2-xAlxGe的电子能量损失函数随光子能量变化的曲线图,由图9看出所,当光子能量小于0.8702 eV时纯Mg2Ge的电子能量损失函数值趋于0,掺入Al后,Mg2Ge能量损失函数发生蓝移,x=0.125时蓝移现象最为明显,最大峰值均有所增大,这是因为杂质电离后产生过剩的游离电子,当入射光子频率达到体系固有振荡频率时,体系振荡强烈,入射光子能量损失增大.
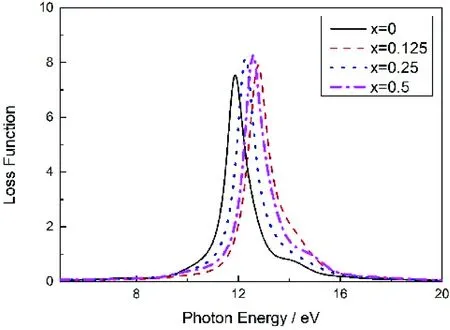
图9 Mg2-xAlxGe的能量损失函数Fig. 9 The electron energy loss functions of Mg2-xAlxGe
4结 论
本文运用第一性原理计算了不同Al掺杂浓度下Mg2-xAlxGe的能带结构、态密度及其光电性质.结果表明,Al掺杂后的Mg2Ge费米能级附近的导带架构发生了改变,变为主要由Al的3p态电子、Ge的4s态电子和Mg的3s、3p态电子组成,呈现出n型导电;静介电常数ε1(0)和折射率n0均增大;吸收光谱发生红移,吸收系数最大值略微减小;光电导率峰值在x=0.125时取得极大值;能量损失函数发生蓝移,最大峰值均有所增大,x=0.125时蓝移现象最为明显.
