一个四代遗传性耳聋家系POU4F3和PDZD7基因突变致病性研究
2020-04-27赵羿耿佳熊文羽张亚娟赵秋棱彭卫华包中伟谭博卢宇程静卜枫啸袁慧军
赵羿耿佳熊文羽张亚娟赵秋棱彭卫华包中伟谭博卢宇程静卜枫啸袁慧军*
1西南大学生命科学学院淡水鱼类资源与生殖发育教育部重点实验室(重庆400715)
2陆军军医大学第一附属医院医学遗传中心(重庆400038)
耳聋是临床上最常见的感觉神经系统缺陷疾病之一。据世界卫生组织统计,全世界超过5%的人口——4.66亿人患有不同程度的听力损失,基因缺陷、机体老化、病毒和细菌感染、药物引起的耳毒性、外伤、暴露于噪音环境等都是听力损失发生的病因,其中约60%由遗传性因素导致。遗传性耳聋根据其是否伴有其他器官异常,可分为综合征型耳聋(syndromic hearing loss,SHL)和非综合征型耳聋(non-syndromic hearing loss,NSHL)两类。非综合征型耳聋根据遗传模式的不同可分为常染色体显性遗传、常染色体隐性遗传、性染色体连锁遗传和线粒体母系遗传。目前遗传性耳聋网站(hereditaryhearingloss.org)已收录遗传性非综合征型耳聋基因119个,常显耳聋基因47个,常隐耳聋基因76个,X连锁遗传基因5个。本研究我们发现了一个涉及四代人的耳聋家系,通过二代测序证实该家系耳聋患者中存在2种致病基因——POU4F3基因和PDZD7基因。这一发现对于耳聋基因诊断具有重要提示作用,同一家系或同一耳聋患者可能存在多种致病基因。随着TGE+MPS模式的广泛使用及全外显子组测序(Whole Exome Sequencing,WES)和全基因组测序(Whole Genome Sequencing,WGS)的成本降低,更高通量的耳聋基因测序技术可以为聋儿家庭提供完整准确的产前诊断指导和专业遗传咨询服务[1]。
1 资料与方法
1.1 家系资料的采集
本家系由西南医院医学遗传中心采集,并通过西南医院伦理委员会认证。该家系来自河南省,编号HL-258。采样小组对先证者及其各亲属成员进行了家系调查,参与研究的家系成员均签署知情同意书,对家系成员进行问卷调查、听力学检查、视觉系统检查、体格检査等。采集各家系成员外周血5ml,提取DNA并-80°C冻存。
1.2 测序及变异位点致病性分析流程
使用课题组自主设计的目标区域捕获试剂盒HHL-785进行检测,该试剂盒覆盖目前已知耳聋基因及文献报道与听觉系统相关基因共785个。目标区域涵盖了785个基因的外显子、外显子上下游50bp及线粒体DNA序列。以GRCh37/hg19为参考序列,测序下机数据应用BWA软件进行原始读长定位,Picard工具进行质控和去重操作,使用GATK和VEP工具进行变异识别和注释,最小等位基因频率(Minor allele frequency,MAF)按0.5%进行变异过滤,过滤后变异应用SIFT、Polyphen-2、LRT和MutationTaster等工具进行蛋白结构和功能预测,应用PhyloP、GERP++工具进行保守性分析。
1.3 Sanger测序验证
针对检出的POU4F3基因和PDZD7基因突变位点设计验证引物,对已采集样本的家系成员进行Sanger测序验证。测序数据质控后,通过Mutation-Surveyor软件进行序列比对,分析检出变异是否符合家系内听力表型和基因型共分离。
1.4 保守性及蛋白三维结构分析
应用ClustalX-2.1对POU4F3和PDZD7在不同物种中的保守性进行分析。应用Swiss-model(https://swissmodel.expasy.org)工具进行三维结构模拟,使用PyMoL软件进行蛋白结构可视化。
2 结果
2.1 HL-258家系资料及听力学特征
HL-258家系通过回访可追溯至4代人,存在耳聋患者5人,参与该研究家系成员共4人。先证者父母Ⅲ-2、Ⅲ-3为非近亲结婚,自家人回忆:在先证者Ⅳ-2幼儿期怀疑其存在听力下降情况,但一直未引起足够重视。先证者Ⅳ-2自述9岁时感知到听力下降,11岁测听双耳中度感音神经性耳聋,全频下降,颞骨CT未见明显异常。先证者父母Ⅲ-2、Ⅲ-3听力正常。Ⅱ-3患者自述30多岁时感到听力下降,71岁测听双耳重度感音神经性耳聋,全频进展性下降。上述四人视觉系统正常,未诉视网膜色素变性(retinitis pigmentosa,RP)相关症状,身体其他系统未见异常,无噪音史及耳毒性药物用药史。

图1 HL-258家系图及听力曲线Fig.1 Pedigree diagram and audiograms of HL-258
2.2 致病基因鉴定
对Ⅱ-3和Ⅳ-2进行测序分析,下机数据通过质控和去重后,使用GATK和VEP进行变异识别和注释。以MAF值<0.5%进行变异位点过滤后,共得到43个候选变异,其中包括4个已报道的良性变异。根据听力表型、VEP Impact注释得分等条件进行筛查后得到两个候选基因变异,Ⅱ-3:POU4F3:NM_002700.3:c.976A>G(p.Arg326Gly)杂合变异,Ⅳ-2:PDZD7:NM_024895.4:c.490C>T(p.Arg164Trp)纯合变异。POU4F3基因该变异为首次报道,该变异gnomAD数据库人群频率为0。致病性预测软件SIFT、Polyphen-2、LRT和 MutationTaster预测皆为有害突变,GERP++和PhyloP预测都为保守性位点。Kim等人在2013年报道了一个POU4F3基因韩国大家系,通过连锁分析鉴定出致病突变POU4F3:c.977G>A(p.Arg326Lys),与本研究Ⅱ-3检出变异为相同氨基酸残基的不同改变[2]。本文PDZD7基因检出变异于2019年9月被韩国学者首次报道,文章共报道两个家系患者与Ⅳ-2具有相同致病突变,听力严重程度与本文情况相似[3]。该变异gnomAD数据库东亚人群频率0.5‰,在3752个正常对照中未检出。SIFT、Polyphen-2、LRT和MutationTaster预测为有害突变,GERP++和PhyloP预测为非保守性位点。
2.3 Sanger测序验证
Sanger测序验证结果表明,Ⅱ-3 POU4F3、PDZD7基因杂合突变,Ⅳ-2 PDZD7基因纯合突变,Ⅲ-2、Ⅲ-3 PDZD7基因杂合突变,Ⅳ-2 、Ⅲ-2、Ⅲ-3三人POU4F3基因为野生型,结合患者Ⅱ-3及Ⅳ-2的听力表型,两基因突变符合基因型与表型共分离,POU4F3、PDZD7基因即为该家系耳聋患者致病基因。该四代耳聋家系存在两种遗传模式,Ⅱ-3一代为常染色体显性遗传。Ⅲ-2由于POU4F3基因为野生型,POU4F3基因突变在该家系未遗传给下一代。Ⅳ-2 PDZD7基因纯合突变遗传自父母双方,为常染色体隐性遗传模式。
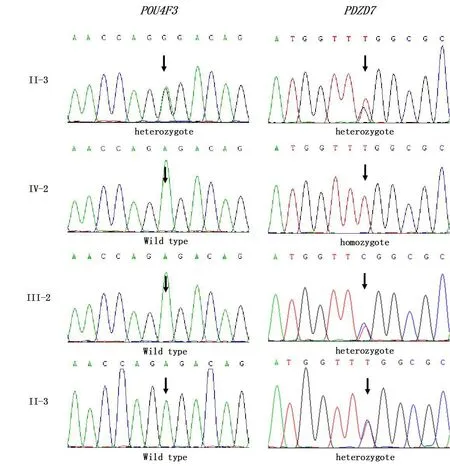
图2 POU4F3、PDZD7 Sanger测序图谱Fig.2 Sanger sequencing chromatograms of the POU4F3 and PDZD7
2.4 保守性及蛋白三维结构模拟分析
将POU4F3氨基酸序列在不同物种中进行比对,Arg326在各物种中高度保守。以2xsd(PDB ID)为模板构建Arg326Gly突变型蛋白进行分析,Arg326位于POU4F3蛋白POU同源结构域的核定位信号(nuclear localization signals,NLSs)上,极性正电荷精氨酸突变为极性不带电荷的甘氨酸后,使Arg326和Pro297之间丧失相互作用,从一定程度上改变了局部二级结构,可能会影响POU4F3蛋白亚细胞定位[4,5]。PDZD7 Arg164在不同物种中高度保守,以2EEH(PDB ID)为模板构建PDZD7突变型蛋白发现,该变异位于PDZD7蛋白第一个PDZ结构域末端,极性精氨酸突变为非极性色氨酸后,局部疏水作用发生改变,使Arg164与Asp82之间产生相互作用,影响局部二级结构的稳定性。
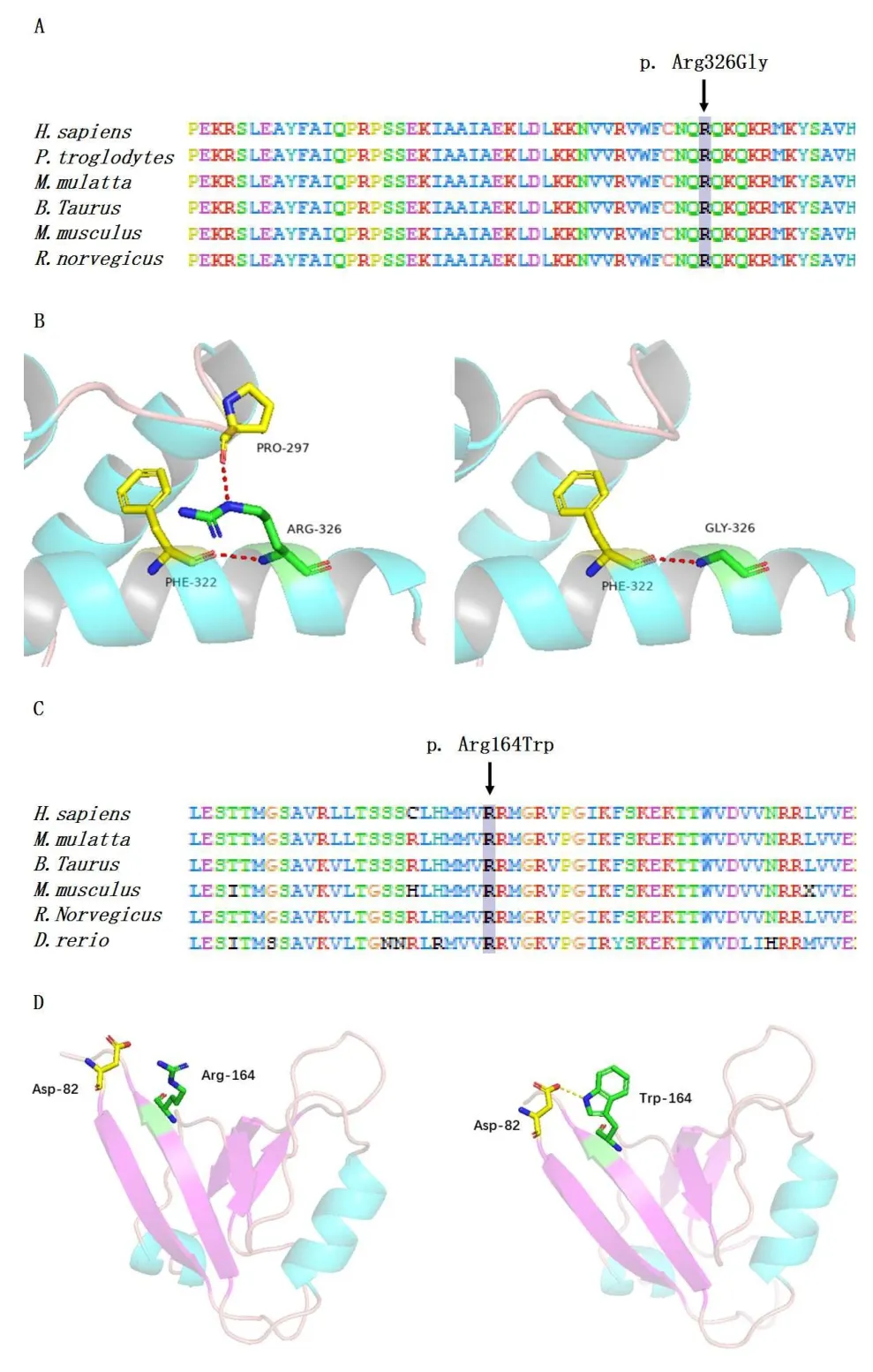
图3 突变位点保守性分析及蛋白三维结构模拟分析。A:POU4F3蛋白Arg326保守性分析;B:POU4F3蛋白野生型&突变型结构比较;C:PDZD7蛋白Arg164保守性分析;D:PDZD7蛋白野生型&突变型结构比较;Fig.3 Conservation and three dimensional structure simulation analysis of the two missense mutations.A:Conservation analysis of the POU4F3 protein;B:Three dimensional structure comparison of wild-type and missense mutant in the POU4F3 protein;C:Conservation analysis of the PDZD7 protein;D:Three dimensional structure comparison of wild-type and missense mutant in the PDZD7 protein
3 讨论
本研究,我们报道了一个具有耳聋家族史的四代家系,不同患者之间听力表型存在明显差异,我们通过二代高通量测序在该家系中鉴定出两个致病基因突 变 位 点 POU4F3:NM_002700.3:c.976A>G(p.Arg326Gly)和 PDZD7:NM_024895.4:c.490C>T(p.Arg164Trp)。Sanger测序验证两突变符合基因型与表型共分离,保守性和蛋白三维结构模拟分析提示两突变干扰蛋白局部二级结构,影响蛋白正常生理功能。
根据美国医学遗传学与基因组学学会(The American College of Medical Genetics and Genomics,ACMG)遗传变异分类标准与指南分析,Ⅱ-3听力情况符合 POU4F3致聋表型(PP4),POU4F3:c.976A>G(p.Arg326Gly)在人群数据库中频率<0.5%(PM2),多种方法预测为致病性变异(PP3),与已知致病性变异POU4F3:NM_002700.3:c.977G>A(p.Arg326Lys)为相同氨基酸残基的不同碱基改变(PM5),但由于无法获取患者Ⅱ-3父母及其患病兄弟的临床资料,故将此变异定义为“可能的疑似致病突变”。本文PDZD7:NM_024895.4:c.490C>T(p.Arg164Trp)变异于2019年9月被韩国学者首次报道,文章报道了2个家系与本文患者Ⅳ-2拥有相同的PDZD7纯合突变,患者均表现为语前中度感音神经性耳聋,在KRGDB(Korean Reference Genome Database)该变异位点频率为0.3%,作者认为PDZD7:p.Arg164Trp为韩国人群高频突变热点,具有奠基者效应(founder effect),根据ACMG指南将该变异位点定义为“可能致病”[3]。本文患者Ⅳ-2同样为中度感音神经性耳聋,但发病年龄晚于韩国病例,推测可能是由于患者之间不同遗传背景的修饰作用所致。
POU4F3基因为POU转录因子IV类家族成员,位于染色体5q31,编码338个氨基酸,包含POU特异性结构域(POU specific domain)和POU同源结构域(POU homeodomain)。POU4F3基因为DFNA15型耳聋致病基因,于1998年Vahava等人在一个五代以色列耳聋大家系中鉴定出来[6],主要临床表型为双侧对称迟发性中-重度进行性感音神经性耳聋。青少年至中年均可发病,全频受损,听力曲线多为平坦型或缓降型。在小鼠中,Pou4f3蛋白主要在内耳毛细胞和前庭毛细胞中强烈表达。Pou4f3-/-小鼠表现出严重的听力损伤和前庭功能异常[7]。目前共报道29个POU4F3致病突变,除以色列(1)、巴西(1)、荷兰(2)外,其余25个致病突变皆为东亚国家——中国(9),日本(13),韩国(3)。Kitano等人认为POU4F3基因(2.5%,15/602)成为日本常染色体显性遗传非综合征型耳聋(ADNSHL)第三大致病基因,仅次于KCNQ4基因(6.6%)和TECTA基因(2.9%)[8]。He等认为POU4F3基因突变也是中国汉族人ADNSHL较为常见的原因[9]。
PDZD7基因位于染色体10q24.3,编码含有PDZ结构域的支架蛋白。PDZD7基因共3个转录本,完整的PDZD7蛋白由三个PDZ结构域、一个HNL结构域(harmonin-N-like domain)和一个富含脯氨酸区域(proline-rich region)组成。在内耳中可以检测到包含前两个PDZ结构域的较短的PDZD7亚型。PDZD7蛋白在内耳中定位于毛细胞机械敏感结构的踝连接(ankle-link)处与其他USH相关蛋白USH2A,GPR98和WHRN形成USH四元复合物[10,11]。2009年,PDZD7基因在一个德国家庭中被确认为非综合征型耳聋DFNB57的致病基因[12],主要临床表型为语前中-重度非进展性感音神经性耳聋,听力曲线多为平坦型或缓降型。PDZD7基因不仅是DFNB57的致病基因,也被认为与Usher综合征2型相关。Usher综合征2型表现为中重度感音神经性耳聋,RP表型约20岁左右出现。Ebermann等认为GPR98和PDZD7双基因突变可引起Usher综合征2型发病,PDZD7基因突变与RP发病相关。USH2A基因纯合突变患者携带PDZD7移码突变会造成RP表型更加严重且发病时间更早[13]。通过ABR评估,Pdzd7-/-KO纯合小鼠表现出先天性极重度耳聋,视网膜电图显示1月龄小鼠视锥、视杆细胞正常。免疫荧光观察到耳蜗内、外毛细胞纤毛束紊乱,影响了与USH2A,GPR98和WHRN组成的USH四元复合物在内耳中的定位[14]。本研究患者Ⅳ-2 PDZD7基因纯合突变,无RP表型,由于患者目前还未到20岁,RP表型还应继续随访。
在本研究中,我们报道了一个具有耳聋家族史的四代家系,使用二代高通量测序鉴定出两种致病变异,扩展了POU4F3基因和PDZD7基因耳聋致病突变谱。在遗传性耳聋家系致病基因鉴定过程中,同一家系检出多种致病基因的情况并不罕见。2014年,本课题组对江西上饶的一个四代耳聋大家系进行致病性分析,该家系遗传方式复杂且患者之间听力表型存在差异,通过高通量测序在不同患者中鉴定出3种致病基因——线粒体12S rRNA A1555G、CDH23和SLC26A4[15]。我们建议在具有耳聋家族史的同一家系中,如果遗传方式复杂且患者之间听力表型存在明显差异,应考虑家系是否可能存在多基因致病的情况,并推荐使用高通量测序技术进行更多耳聋致病基因检测和诊断,以提高致病基因检出率,减少漏诊。随着近年来TGE+MPS模式的广泛应用和WES、WGS成本的降低,使得致病基因快速筛查和更多耳聋新基因的发现成为可能,从而为耳聋家庭生育后代时提供更加专业的遗传咨询和生育指导,降低聋儿出生率,减轻家庭及社会的负担。
