多卤代吡啶选择性合成醚类的反应*
2020-03-31鲍建设姚武冰
鲍建设,张 斌,姚武冰
(医药化工与材料工程学院台州学院,浙江 台州 318000)
吡啶类化合物具有广泛的生物活性和药理活性,是天然产中的重要骨架之一[1](图1),广泛应用于药物[2]、荧光探针[3]、受体配体[4]、共轭有机材料[5]等研究领域。

图1 含有吡啶官能团的天然产物
吡啶衍生物的合成方法主要通过为下列两种方法:(1)过渡金属催化的吡啶的C-H键的官能化反应[6];(2)卤代吡啶经偶联反应制得相应的化合物[7]。过渡金属催化的C-H键的官能团化反应具有原子经济性,是非常具有研究前景的合成方法,但这类反应通常有很多限制,如反应温度较高,催化剂受限等因素。相比而言,卤代吡啶的C-X键具有更高的活性,最近十几年中,已有文献报道了多卤代吡啶的区域选择性偶联反应来构建不同的吡啶衍生物[8-14]。但是多卤代吡啶参与的选择性C-O[15]反应和C-S[16]反应的报道的较少,而且反应涉及到金属配体,高温,不易操作等缺点,因此我们开发了无金属,温和条件下的多卤代吡啶与苯酚和硫酚的选择性生成C-O和C-S键的反应(图2)。

图2多卤代吡啶与苯酚和硫酚的选择性反应
Fig.2 2-aryloxyation and 2-thiolation of 2,x-dihalopyridines
1 实 验
1.1 仪器与试剂
核磁共振谱图采用Bruker AV400型核磁共振仪测定(CDCl3为溶剂,TMS为内标),质谱采用Waters Synapt-G2-Si飞行时间质谱仪测定。反应中所使用的试剂都直接从商业途径购买并直接使用,未经任何处理。
1.2 化合物3a~3i和5a~5i的合成以及表征
在25 mL圆底烧瓶中加入卤代吡啶(0.5 mmol),苯酚(1.0 mmol),KOH(1.0 mmol),DMSO(5 ml),在50 ℃下空气中搅拌6 h。TCL跟踪反应,反应完后,用CH2Cl2(20 mL)和饱和NaCl 溶液(20 mL)对反应液进行萃取,分出有机相,用无水硫酸钠干燥,过滤,蒸去有机溶剂,浓缩物用经柱层析分离得到目标产物3a~3i,经核磁分析及文献[15]对照,产率36%~95%。
在25 mL圆底烧瓶中加入卤代吡啶(0.5 mmol),苯硫酚(1.0 mmol),K2CO3(1.0 mmol),DMSO/H2O(2:1)(5 mL),在30 ℃下空气中搅拌3 h。TCL跟踪反应,反应完后,用CH2Cl2(20 mL)和饱和NaCl 溶液(20 mL)对反应液进行萃取,分出有机相,用无水硫酸钠干燥,过滤,蒸去有机溶剂,浓缩物用经柱层析分离得到目标产物5a~5i,经核磁分析及文献[16]对照,产率72%~97%。
3a: pale yellow liquid, 收率94%;1H NMR(CDCl3, 400 MHz): 8.20(d, 1H), 7.45(t, 2H), 7.29(t, 1H), 7.09(d, 2H), 6.98(d, 1H), 6.82(dd, 1H);13C NMR(CDCl3, 100 MHz): 166.3, 153.7, 151.2, 143.1, 130.7, 126.3, 121.1, 115.9, 112.1; MS(ESI): 249.9[M+H]+。
3b: yellow solid, 收率95%, m.p.63.1~64.7 ℃;1H NMR(CDCl3, 400 MHz): 8.17(d, 1H), 7.24(d, 2H), 6.96(d, 3H), 6.80(dd, 1H), 2.38(s, 3H);13C NMR(CDCl3, 100 MHz): 166.5, 151.3, 151.2, 143.2, 136.0, 131.1, 120.8, 115.7, 111.9, 21.1; MS(ESI): 263.9[M+H]+。
3c: yellow solid, 收率92%, m.p.64.7~66.1 ℃;1H NMR(CDCl3, 400 MHz): 8.18(d, 1H), 6.94~7.03(m, 5H), 6.79(dd, 1H), 3.83(s, 3H);13C NMR(CDCl3, 100 MHz): 166.9, 157.7, 151.1, 146.8, 143.1, 122.1, 115.6, 115.4, 111.7, 55.9; MS(ESI): 280.0[M+H]+。
3d: pale yellow solid, 收率82%, m.p.67.4~68.8 ℃;1H NMR(CDCl3, 400 MHz): 8.22(d, 1H), 7.39~7.43(m, 2H), 6.98~7.06(m, 3H), 6.82(dd, 1H);13C NMR(CDCl3, 100 MHz): 165.9, 152.2, 151.4, 143.2, 131.6, 130.7, 122.4, 115.9, 112.0; MS(ESI): 283.9[M+H]+。
3e: pale yellow liquid, 收率92%;1H NMR(CDCl3, 400 MHz): 8.20(d, 1H), 7.72(dd, 1H); 7.19(d, 2H), 6.99~7.01(m, 2H), 6.79(d, 1H), 2.35(s, 3H);13C NMR(CDCl3, 100 MHz): 163.1, 151.6, 148.5, 142.1, 134.9, 130.5, 121.3, 113.4, 113.1, 21.1; MS(ESI):264.1[M+H]+。
3f: yellow liquid, 收率95%;1H NMR(CDCl3, 400 MHz): 8.05(dd, 1H), 7.90(dd, 1H), 7.40(t, 2H), 7.22(t, 1H), 7.15(d, 2H), 6.86(q, 1H);13C NMR(CDCl3, 100 MHz): 160.1, 153.9, 146.2, 142.8, 129.8, 125.3, 121.6, 119.8, 108.0; MS(ESI): 249.9[M+H]+。
3g: yellow solid, 收率80%;1H NMR(CDCl3, 400 MHz): 8.00(d, 1H), 7.92(d, 1H), 7.41(t, 2H), 7.24(m, 1H), 7.13(m, 2H);13C NMR(CDCl3, 100 MHz): 158.5.1, 153.5, 144.3, 141.9, 129.7, 125.7, 125.4, 121.4, 107.9; MS(ESI): 283.9[M+H]+。
3h: colorless liquid, 收率82%;1H NMR(CDCl3, 400 MHz): 7.95(d, 1H), 7.72(d, 1H), 7.20(d, 2H), 7.02(d, 2H), 2.35(s, 3H);13C NMR(CDCl3, 100 MHz): 158.1, 151.2, 143.7, 138.9, 135.2, 130.4, 125.5, 121.4, 119.6, 21.1; MS(EI): 252.1(M+)。
3i: colorless liquid, 收率36%;1H NMR(CDCl3, 400 MHz): 8.26(s, 1H), 7.96(d, 1H), 7.24(d, 2H), 7.05(d, 2H), 2.38(s, 3H);13C NMR(CDCl3, 100 MHz): 161.8, 150.8, 142.9, 136.5, 135.8, 130.6, 124.5, 122.2(m), 121.6, 119.5, 21.2; MS(EI): 287.1(M+)。
5a: yellow liquid, 收率97%;1H NMR(CDCl3, 400 MHz): 8.05(d, 1H), 7.42(d, 2H), 7.27(d, 2H), 7.03(s, 1H), 6.84~6.85(m, 1H), 2.41(s, 3H);13C NMR(CDCl3, 100 MHz): 154.6, 149.3, 142.5, 140.9, 135.6, 131.2, 124.7, 123.8, 119.7, 21.6; MS(EI): 279.0(M+)。
5b: yellow liquid, 收率94%;1H NMR(CDCl3, 400 MHz): 8.06(d, 1H), 7.47(d, 2H), 7.00(d, 3H), 6.84(d, 1H), 3.87(s, 3H);13C NMR(CDCl3, 100 MHz): 161.6, 155.5, 149.2, 142.4, 137.6, 123.6, 119.5, 118.5, 116.1, 55.7; MS(EI): 297.0(M+)。
5c: yellow liquid, 收率89%;1H NMR(CDCl3, 400 MHz): 8.11(dd, 1H), 7.26(s, 1H), 7.04(dt, 1H), 2.96(t, 2H), 1.71(m, 2H), 1.49(m, 2H), 0.97(t, 3H);13C NMR(CDCl3, 100 MHz): 153.1, 149.3, 142.6, 123.9, 120.0, 30.7, 30.5, 22.2, 13.8; MS(EI): 245.0(M+)。
5d: yellow solid, 收率82%;1H NMR(CDCl3, 400 MHz): 8.10(d, 1H), 7.47(q, 4H), 7.06(s, 1H), 6.87(d, 1H);13C NMR(CDCl3, 100 MHz): 153.1, 149.7, 142.8, 137.0, 136.8, 130.7, 127.1, 124.2, 120.0; MS(ESI): 299.9[M+H]+。
5e: white solid, 收率89%;1H NMR(CDCl3, 400 MHz): 8.24(dd, 1H), 7.71(dd, 1H), 7.48(dd, 2H), 6.96(d, 2H), 6.86(dd, 1H), 3.64(s, 3H);13C NMR(CDCl3, 100 MHz): 160.8, 160.0, 148.2, 139.8, 137.6, 120.8, 118.2, 115.2, 55.6; MS(EI): 296.0(M+)。
5f: white solid, 收率96%;1H NMR(CDCl3, 400 MHz): 8.45(d, 1H), 7.49(m, 3H), 7.24(d, 2H), 6.73(d, 1H), 2.39(s, 3H);13C NMR(CDCl3, 100 MHz): 161.2, 150.4, 140.1, 139.4, 135.5, 130.9, 126.9, 122.4, 116.5, 21.6; MS(EI): 280.0(M+)。
5g: yellow liquid, 收率72%;1H NMR(CDCl3, 400 MHz): 8.16(d, 1H), 7.58(d, 1H), 7.42(d, 3H), 7.23(d, 2H), 2.39(s, 3H);13C NMR(CDCl3, 100 MHz): 156.6, 146.5, 139.9, 135.9, 135.8, 130.4, 128.6, 128.1, 125.7, 21.7; MS(EI): 270.1(M+)。
5h: yellow solid, 收率91%;1H NMR(CDCl3, 400 MHz): 8.46(d, 1H), 7.47(d, 1H), 7.15(m, 2H), 6.87(m, 2H), 2.39(s, 3H), 2.35(s, 3H);13C NMR(CDCl3, 100 MHz): 153.8, 149.2, 142.8, 142.1, 136.9, 132.3, 128.4, 123.6, 123.2, 119.1, 21.3, 20.5; MS(ESI): 293.9[M+H]+。
5i: yellow solid, 收率88%;1H NMR(CDCl3, 400 MHz): 8.19(d, 1H), 7.46(q, 1H), 7.42(d, 2H), 7.23(d, 2H);13C NMR(CDCl3, 100 MHz): 157.8, 146.7, 139.6, 138.9, 135.5, 130.2, 130.2, 127.8, 126.1, 117.7, 21.4; MS(ESI): 313.9[M+H]+。
2 结果与讨论
2.1 选择性C-O反应的条件优化
首先将2,4-二溴吡啶(0.5 mmol)、苯酚(0.6 mmol)、K2CO3(1.0 mmol)和DMSO(5 mL)放入反应瓶中,30 ℃下空气中搅拌6 h,将反应产物经过后处理,得到目标产物3a(表1),其产率为45%(Entry 1)。同样的条件下,以K2CO3为碱,筛选了不同的溶剂,如THF、MeOH、DMF,结果显示都没有DMSO作为溶剂效果好(Entry 2~4)。在选择碱的时候,我们发现,碱性越强,产率越高,因此选择了KOH(Entry 5~6)。同时,我们将苯酚的量增加到二溴吡啶的2倍时,收率有明显的提升(Entry 7)。温度和时间对反应的影响也很大,温度过低时产物转化率不高,过高时反应的选择性变差,有副产物生成,产率降低,反应时间长也会导致副产物的生成,收率下降(Entry 8~10)。最终,我们选择的最佳反应条件是2,4-二溴吡啶(118.4 mg,0.5 mmol),苯酚(94.1 mg,0.6 mmol),KOH(56 mg,1.0 mmol),DMSO(5 mL),在50 ℃空气中搅拌6 h。


表1 反应条件优化a
aReaction conditions: 2,4-Dibromopyridine(0.5 mmol), phenol(0.6 mmol), base(1.0 mmol), solvent(5 mL), 6 h;bphenol(1.0 mmol);cReaction time 12 h。
2.2 选择性C-O反应的底物拓展
在上述最佳反应条件下,我们将多卤代吡啶与各种苯酚反应,合成了2位苯氧基取代的吡啶衍生物(表2)。从表2可以得出,2,4-二溴吡啶与含供电子基团的苯酚反应时,产物的收率和选择性都非常好,达到92%~94%(3a,3b,3c)。但当含吸电子基团的对氯苯酚参与反应时,收率低至82%(3d)。此外,2,3-二溴吡啶,2,5-二溴吡啶与苯酚反应都能得到非常高区域性和高收率的产物(3e,3f)。值得注意的是,当吡啶2位上的溴原子换成氯原子时,依旧有很好的选择性和收率(3g,3h),但当吡啶环上有强吸电子基团三氟甲基时,选择性明显变差,目标产物收率只有36%(3i)。

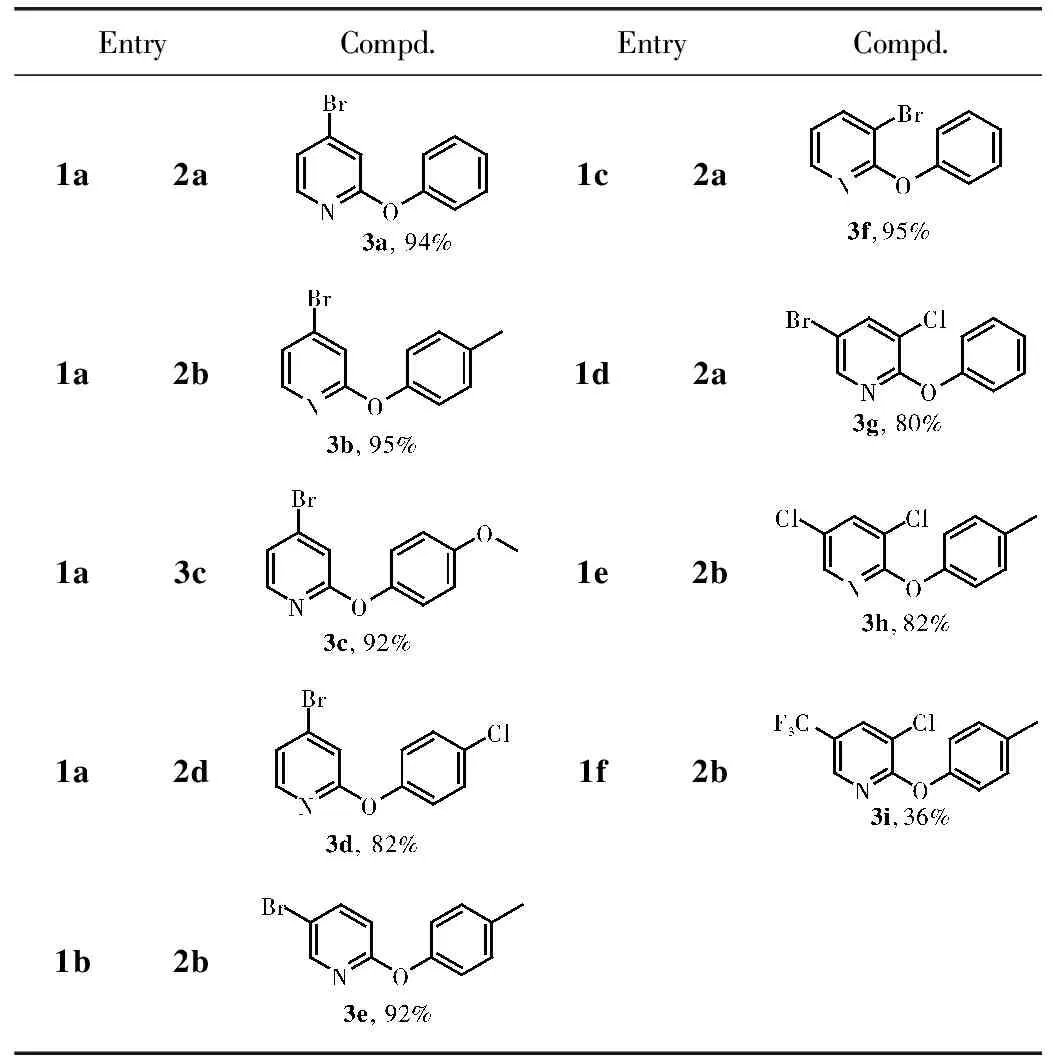
表2 2-苯氧基-溴代吡啶的合成a
aReaction conditions: halopyridines(0.5 mmol), phenols(1.0 mmol), base(1.0 mmol), solvent(5 mL), in air at 50 ℃, 6 h。
2.3 选择性C-S反应的条件优化


表3 反应条件优化a
aReaction conditions: halopyridines(0.5 mmol), thiols(0.6 mmol), base(1.0 mmol), solvent(5 mL), 3 h;bDMSO/H2O(V:V=4:1);cDMSO/H2O(V:V=2:1);dDMSO/H2O(V:V=1:1);ehalopyridines(0.5 mmol), thiols(1.0 mmol)。
在二溴吡啶与苯酚的选择性反应的基础上,我们又研究了二溴吡啶与苯硫酚的选择性生成C-S键反应(表3),我们将二溴吡啶与苯酚的最优条件用于对甲苯硫酚时,发现选择性并不理想,副产物较多,目标产物的收率只有35%(Entry 1)。考虑到KOH的碱性太强导致选择性反应不理想,收率也不高,因此我们筛选了碱性弱一点的碱Cs2CO3、K2CO3、K3PO4,当选择K2CO3时,区域选择性有明显的提高,副产物也明显减少,因此目标产物收率升至65%(Entry 2~4)。此外,我们在探索条件中还发现该反应在有水相参与的情况下任然有较高的收率,发现以DMSO/H2O(V:V=2:1)做溶剂时,反应仍然有非常好的选择性和收率,最高达到97%(Entry 5~8)。最终我们发现的最优条件是以2,4-二溴吡啶(118.4 mg,0.5 mmol)、对甲基苯硫酚(124.1 mg,1.0 mmol)、K2CO3(138.3 mg,1.0 mmol)和DMSO/H2O(V:V=2:1)5 mL,在30 ℃下空气中搅拌3 h,反应结束收到目标产物5a收率为97%。
2.4 选择性C-S反应的底物拓展
在上述最佳反应条件下,多卤代吡啶与各种苯硫酚反应,合成了2位苯氧基取代的吡啶(表4)。在表中可以看出,2,4-二溴吡啶、2,5-二溴吡啶、2,3-二溴吡啶、2,3-二溴-5-氯吡啶与含供电子基团苯硫酚反应,都能高选择性和高产率地得到了2位苯硫基取代的吡啶产物(5a,5b,5e,5f,5i),但与含吸电子基团的苯硫酚反应时,收率有所降低(5c,5d)。当2,3,5-三氯吡啶参与反应时,产率降低,这是由于C-Cl键相较于C-Br键活性低(5g),值得注意的是尤其是当含有位阻基团的2,4-二甲基-苯硫酚参与反应时,仍然有较高的收率(5h)。

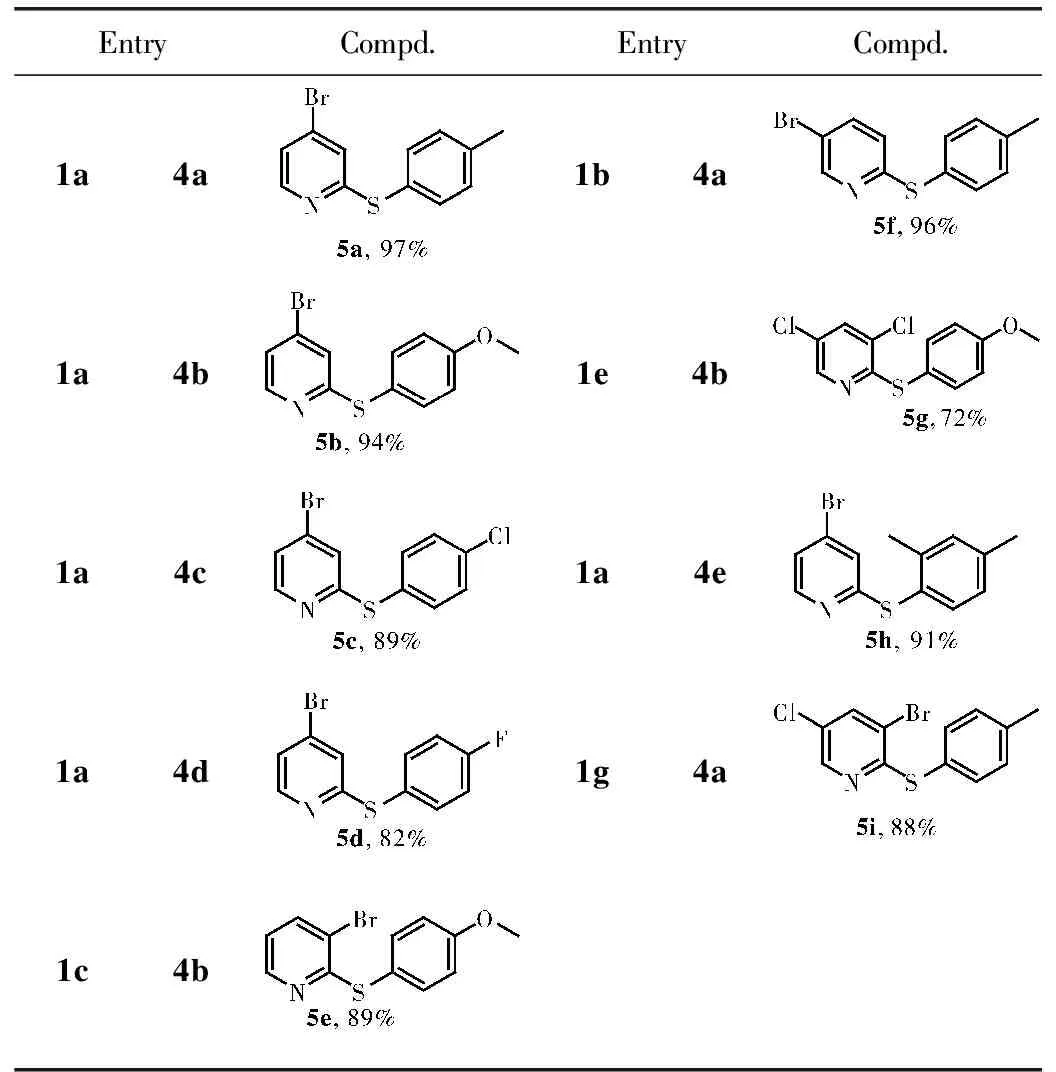
表4 2-苯硫基-溴代吡啶的合成a
aReaction conditions: halopyridines(0.5 mmol), thiols(1.0 mmol), base(1.0 mmol), solvent(5 mL), in air at 30 ℃, 3 h。
3 结 论
最终,我们开发了一种无金属催化,在温和条件下的进行多卤代吡啶与苯酚和硫酚的选择性生成C-O和C-S键的反应的方法。该方法具有很好的收率和区域选择性,是一种制备吡啶衍生物的有效方法。
