中年男性,进行性行动迟缓2年——早发型帕金森病
2020-03-16黄招君周美鸿洪道俊
黄招君 周美鸿 洪道俊
1 资料
患者,男,39岁,因“进行性行动迟缓2年”于2020年6月20日就诊于南昌大学第一附属医院神经科。
患者于2年前开始感乏力,自述体力不如从前,行动能力下降。数月后出现走路缓慢,主要表现为跟不上同行的人,同时也合并上肢动作缓慢,表现为刷牙费力,持筷进食不灵活。约1年前家人发现其走路勾背,起步有时困难,有时往一侧歪斜。上述症状进行性加重,但患者未重视也没有就诊。最近半年患者右手出现细微震颤,感觉肢体僵硬感,以右侧肢体为重。同时伴随起步困难,蹲下站起困难,走路时下肢拖曳,有时前冲步态,易向右侧偏斜,右上肢摆臂动作减少。患者有时头晕,无明确视物旋转,和体位无明显关系,无恶心呕吐。无头痛,无心悸胸闷。饮食尚可,无胃肠不适。睡眠尚可,家人未发现其夜间喊叫,无睡眠相关损伤事件。大便干结,2~3天1次,小便功能尚可。
既往史及家族史:患者约1岁半开始行走,近3岁开口说话,语言少,完整句子不能很好表达,反应或者做事较同龄人慢半拍,简单事情可跟同龄人同等程度完成,复杂问题解决能力稍差,读书成绩不理想。无其他特殊疾病史,无药物或毒物接触史。父母身体健康,非近亲结婚,有两个姐姐 (图1A),其中有二姐,41岁,从小智力比同龄人落后,走路时会左右摇晃,症状和患者部分类似,但智力更差。
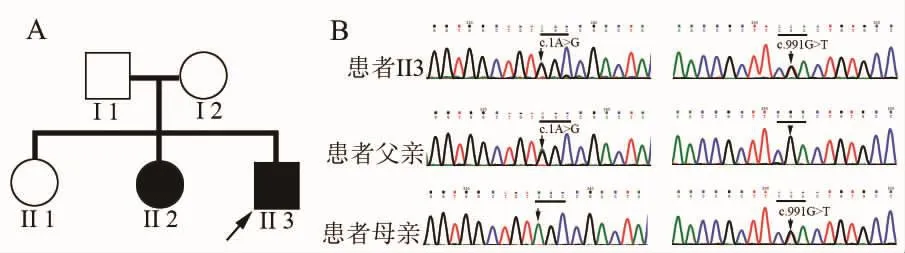
图1 患者家系图及基因变异 家系图显示患者(II3)和其姐姐(II2)有类似症状。基因测序显示患者携带c.1A>G和c.991G>T符合杂合突变,其中c.1A>G来自父亲,c.991G>T来自母亲,符合家系共分离现象。
体格检查:体温:36.5℃,脉搏:109 次/min,呼吸:16 次/min,血压:104 mmHg/77 mmHg,心律齐,叩诊心界不大,各瓣膜区听诊未闻及杂音。神志清楚,面具脸,言语含糊。简易智能精神状态检查量表评分(MMSE)23分,蒙特利尔认知评估量表(MoCA)21分。双侧瞳孔等大等圆,直径约3.0 mm,直接及间接对光反射灵敏。双眼睑闭合力可,双侧眼球向各个方向运动正常,未及复视和眼震。角膜反射正常,双侧面部痛温觉无减退。双侧颞肌、咀嚼肌对称,咬肌力弱。双侧额纹对等,鼻唇沟对称。双侧听力粗测正常。悬雍垂居中,双侧软腭上抬有力,咽反射存在。双侧转颈、耸肩正常。伸舌居中,无舌肌萎缩及震颤。颈屈肌力5级,转颈肌张力显著增高。右手出现静止震颤,频率约4~6次/s,四肢未见明显肌萎缩,双上肢肌张力轻度齿轮样增高,右下肢肌张力增高,右侧肢体肌力5-级,左侧肢体肌力5级。双侧指鼻试验能完成,可见意向性震颤,双侧轮替试验和跟膝胫试验欠稳准。起步困难,前冲小碎步态。并步直立试验睁眼不稳,闭目完全不能站立。双上肢腱反射活跃,双下肢腱反射引出。掌颌反射和吸吮反射阴性,右上肢Hoffmann's征阳性,双下肢病理征阴性。脑膜刺激征阴性。自主神经功能检查正常。
辅助检查:血、尿、便常规正常,肝肾功能、血糖、血脂、电解质、肌酶谱、同型半胱氨酸均正常,铜蓝蛋白正常,甲状腺功能正常,自身免疫相关抗体阴性。普通心电图未见明显异常。胸部CT正常,超声心动图示心脏构筑结构和瓣膜功能正常。腹部彩超:胆囊息肉,肝、胰、脾、双肾彩超未见异常。患者头颅磁共振显示小脑皮质萎缩(图2A、B)。患者二姐头颅磁共振显示小脑皮质轻微萎缩(图2C、D)。
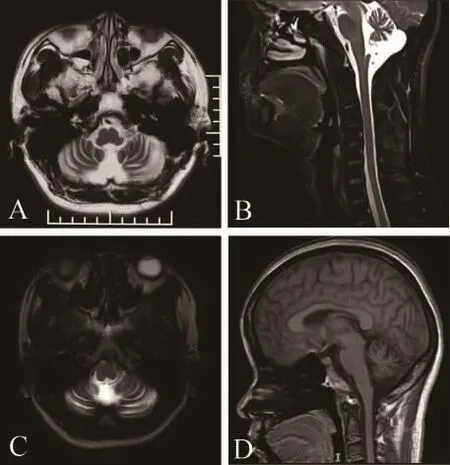
图2 PLA2G6基因突变患者头颅核磁 患者头颅磁共振显示小脑皮质萎缩(A、B)。患者二姐头颅磁共振显示小脑皮质轻微萎缩(C、D)。
基因检查:二代测序显示磷脂酶A2第Ⅵ型基因(PLA2G6)存在两处杂合变异:一个为c.1A>G,直接影响翻译起始密码,该位点为文献已经报道致病突变;另一个为c.991G>T,导致氨基酸改变p.D331Y,该位点也为文献已经报道致病突变。家系验证显示c.1A>G来自父亲,c.991G>T来自母亲,患病的二姐同时携带c.1A>G和c.991G>T突变,因此上述基因变异符合家系共分离现象(图1B)。综上所述,该患者临床基因诊断为PLA2G6基因相关早发型帕金森病14型(PARK14)。
临床疑诊帕金森病(Parkinson disease,PD)后,即给予该患者美多芭125 mg每天3次,餐前1 h口服,盐酸普拉克索0.125 mg每天3次口服,患者服药后3 d症状明显改善,统一帕金森综合评分量表(UPDRS)III评分从服药前31分,减少为18分。基因确诊后,调整为美多芭125 mg每天3次,餐前1 h口服,盐酸普拉克索0.25 mg每天3次口服,目前患者运动能力较前显著改善,能正常生活和工作。
2 讨论
PD是一种常见的中老神经系统退行性疾病,其临床主要表现为运动迟缓、肌强直、静止性震颤、直立不稳等运动症状,以及嗅觉减退、睡眠障碍、情感障碍等非运动症状[1]。PD的平均发病年龄为60岁左右,我国65岁以上人群PD的患病率大约1.7%,因而经典的PD并不属于罕见病范畴[2]。然而,2018年5月22日国家卫健委发布的第一批121种罕见病目录中,PD(青年型、早发型)被列为罕见病。大部分经典的PD患者为散发病例,约5%~10%的PD患者存在潜在基因变异,而其中仅有3.7%~16.6%的患者能够被发现明确的致病基因,此类患者发病年龄通常小于45岁,且临床症状有时不典型[3]。基于此我们这例PLA2G6基因突变导致的早发型PD14型可以被认为罕见病。
PLA2G6基因相关性PD具有高度的临床异质性[4],多数患者在青少年期起病,在幼儿期可以精神运动发育迟缓或者心理障碍为首发症状[5],但在此阶段很难被识别和诊断,我们该例患者就表现为轻度的运动发育迟缓和智力障碍,在很长一段时间内被家属忽略。随着病情发展,患者可逐渐出现运动迟缓、肌强直、静止性震颤等PD症状[6],我们该例患者到中年逐渐出现上述症状,无论临床表现,还是查体都提示存在帕金森综合征的临床表型,故临床疑诊了早发型PD。此外,该例患者表现为双上肢腱反射增强、上肢病理征阳性,提示合并锥体束损害;共济障碍合并小脑萎缩,提示小脑系统及其联系纤维受累[7]。PLA2G6基因相关性PD可以合并上述椎体束和小脑等多系统受累,这也是其他早发型PD患者常常合并存在的临床表征[8]。
隐性遗传的PLA2G6基因突变不仅可以导致PARK14,也可以与肌张力障碍-帕金森综合征(DP)[9]、脑铁沉积变性病(NBIA)[10]、婴儿神经轴索变性(iNAD)[11]、精神分裂症[12]、Ⅰ型糖尿病[13]等疾病相关。早发型PD的临床特点,除了发病年龄小于45岁是一个重要特征之外,临床常常合并多系统神经症状,特别是肌张力障碍。因此,当PD患者合并肌张力障碍时,除了考虑药物治疗反应之外,需要考虑到该PD患者可能存在基因突变的可能[14]。我们该例患者尽管根据其核心症状被诊断为PARK14,但是同时合并存在自幼出现的精神运动发运迟缓,以及小脑症状和锥体束征,这些临床症状横跨PARK14、DP、iNAD等几个症候群,也提示PLA2G6基因突变患者的临床表型在不同亚型之间有一定的重叠性,因此可以其最核心的临床表型将其归类为PARK14,也可以将其认为PLA2G6相关神经系统变性疾病的独立类型[15]。PLA2G6突变所致临床异质性大,使其临床诊断十分困难,极易造成漏诊和误诊。患者的锥体外系、锥体束征、认知障碍症状需注意和脊髓小脑型共济失调、脑瘫及多巴反应性肌张力障碍等鉴别。如果神经科医生对PD或者帕金森综合征的认识仍停留在老年患病、运动迟缓、肌强直、静止性震颤等典型表现层面,患者很难得到正确诊治。
PLA2G6相关神经系统变性疾病是一组常染色体隐性遗传性疾病,目前报道的患者数并不多,因此临床型和基因型之间的关系尚不明确[16]。我们该例患者c.1A>G突变来自其父亲,导致起始密码子不能正常启动蛋白合成,文献报道该突变与婴儿神经轴索变性患者相关[17];c.991G>T突变来自其母亲,导致p.D331Y变异,文献报告该突变与早发型PD患者相关[18];这两个突变在患者及其患病的姐姐体内呈现复合杂合突变,因此这两个突变的致病性基本可以确定。c.1A>G突变和婴儿神经轴索变性相关,也许正是此家系患者存在自幼出现的精神运动发育迟缓表型的原因。进一步的临床型和基因型之间的关系,需要后续更多的患者进行确认。
PARK14治疗初期给予多巴胺能药物可以产生戏剧性反应,但维持时间短暂,且在治疗期间可能出现运动并发症,但多巴胺能药物治疗仍为首选方案[19-20]。对PARK14患者行深部电刺激、经颅磁刺激等神经调控治疗是一种可选治疗模式,但该治疗只适用于部分人群,且PARK14异质性显著,不同亚型刺激位点的选择及模式尚需进一步研究。万志荣等[21]曾报告1例PARK14患者行深部电刺激手术治疗,术后患者病情稍有改善,表现为异动减少,但步态不稳、行动迟缓无明显改善,总体疗效欠佳。我们该患者服用美多芭后3天症状明显改善,但结合既往报告,该患者长期转归还需继续跟踪随访。有效的治疗方案应从PD的发病机制中寻求新的突破口,既往体内外试验提出PD可能与内质网应激、钙稳态异常、线粒体功能障碍、活性氧增加和凋亡等一系列级联事件有关,偶氮酰胺可通过保护PLA2G6的D331Y突变的多巴胺能神经元免受内质网应激诱导的氧化损伤,起到神经保护作用,所以偶氮酰胺是一种治疗PD潜在候选药物,是否对PLA2G6突变导致的PD具有潜在治疗作用值得进一步观察[22]。
3 点评
目前PD的病因及发病机制尚不完全明确,可能与老化、环境、遗传等因素相关。随着分子生物学技术的发展,单基因遗传因素在PD发病机制中的作用越来越受到关注[23]。到目前为止被克隆的PD相关单基因超过20个[24],如与常染色体显性遗传性PD相关的致病基因有SNCA、LRRK2、UCHL1、POLG、DNAJC13、TMEM230、VPS35、HTRA 2、E1F4G1、CHCHD2、GIGYF2、GBA、LRP10、NOCTH2NLC 等,与常染色体隐性遗传PD相关的致病基因有PARKIN、PINK1、PARK7、DJ-1、PLA2G6、ATP13A2、FBXO7、DNAJC6、SYNJ1等,以及与X染色体连锁遗传的致病基因有ATP6AP2。有研究显示在45岁以下的早发型PD患者人群中,这些基因突变大概占到不足50%的患者,提示还有很多潜在的致病基因没有发现。
单基因遗传性PD患者通常具有一些独特的临床表型[25],包括:①发病年龄早,通常小于 45岁,多在 30~40岁发病,属于早发型或者青年型PD范畴;②早期运动症状较轻,且运动症状不典型,起病不对称;③部分患者在运动症状出现前,已经存在其他系统症状;④局限性肌张力障碍常见,特别是肢体远端的肌张力障碍常见;⑤常伴有肢体的腱反射活跃或亢进,提示锥体束损害;⑥部分患者可以伴随认知障碍、癫痫、小脑共济失调等多系统症状;⑦部分患者临床症状有波动性,常见疲劳现象,且早期对小剂量多巴制剂敏感;⑧头颅影像学检查一般无异常改变。因此,在临床上遇到45岁以前起病,特别是伴随肌张力障碍或者其他多系统受累的PD患者,要警惕遗传因素在致病机制中起的作用,及时建议患者进行基因检测,这对患者的长期管理具有积极意义。
