南天竹总生物碱提取纯化工艺研究*
2020-03-04陈祥云彭财英潘玲玲卢健陈圣加舒积成
★ 陈祥云 彭财英 潘玲玲 卢健 陈圣加 舒积成*
(江西中医药大学 南昌 30004)
南天竹(Nandina domestica),小檗科(Berberidaceae)南天竹属(Nandina)小灌木,别名:南天竺,红杷子,天烛子,红枸子,钻石黄,天竹,兰竹等[1]。主要分布在湖北、江苏、浙江、安徽、江西、山东、广西、云南、四川等省,具有观赏、生态、药用等多种价值[2]。南天竹的次生代谢产物为生物碱、黄酮、挥发油类等,而生物碱成分为主要的活性成分,具有降压,抗痉挛,抗菌,止咳平喘,解毒等作用[3]。文献及本课题组研究发现,南天竹提取物对抗肿瘤药三氧化二砷具有较好的减毒作用[4-5]。另外,本课题组前期研究结果显示,南天竹总生物碱对三氧化二砷所致心毒具有明显拮抗作用[6]。因此,有必要对南天竹总生物碱提取纯化工艺进行优化,为南天竹的进一步开发和利用提供参考。
1 仪器与材料
1.1 仪器 UV-2550 型紫外分光光度计;电子分析天平(北京塞多利斯仪器有限公司);恒温水浴锅(杭州庚雨仪器有限公司,HH-ZK2 型);R501 旋转蒸发器(杭州庚雨仪器有限公司);KQ-250DB 型数控超声波清洗器(昆山市超声仪器有限公司);MH-1000 型可调式电热套(北京科伟永兴仪器有限公司);SHB-Ⅲ循环水式多用真空泵(杭州庚雨仪器有限公司);低温冷却液循环泵(杭州康雨仪器有限公司,DLSB-5/20 型);电热恒温鼓风干燥箱(上海精宏实验设备有限公司、太仓精宏仪器设备,DHG-9070A 型)。
1.2 试剂与试药 南天竹购于江苏苏州,经江西中医药大学付小梅教授鉴定;盐酸小檗碱对照品(北京赛百草科技有限公司,纯度:≧98%,批号:633658);D101、HPD-722、AB-8、NKA- Ⅱ、NKA-9、ADS-17 型大孔吸附树脂、732 阳离子树脂(河北沧州宝恩化工有限公司);溴甲酚绿(上海馨晟试化工科技有限公司,批号:20160908);溴百里香酚蓝(上海馨晟试化工科技有限公司,批号:20160602);其它化学试剂均为分析纯。
2 方法与结果
2.1 南天竹中总生物碱提取条件研究
2.1.1 总生物碱的含量测定方法的确定 对照品溶液的制备 精密称取盐酸小檗碱对照品约12.8mg,置50mL 容量瓶中,加甲醇溶解并稀释至刻度,摇匀,精密量取1mL 至25mL 容量瓶中,加甲醇稀释至刻度,配成浓度为10.24µg·mL-1的盐酸小檗碱对照品溶液。
酸性染料的配制 溴甲酚绿:称取0.1g 溴甲酚绿粉末于研钵中,加入2.8mL 的NaOH,研磨至粉末充分溶解后,加入200mL 的蒸馏水稀释,移入具塞的广口瓶中备用。溴百里香酚蓝:称取0.1g 溴百里香酚蓝粉末于研钵中,加入3.2mL 的NaOH,研磨至粉末充分溶解后,加入200mL 的蒸馏水稀释,移入具塞的广口瓶中备用。
酸性染料比色法 精密吸取一定量的供试品溶液,用pH=5.0 的磷酸二氢钾缓冲液4mL 溶解,转移至含有酸性染料3mL 的分液漏斗中,摇匀静置30min,加入氯仿5mL,震摇静置10min,取氯仿层,萃取两次,合并。
测试波长确定 取1mL 对照品溶液分别用2种显色剂,显色后,经5mL 氯仿分2 次萃取,氯仿定容至25mL,氯仿萃取液作为2 种测试液,以无水甲醇为空白对照,在200~600nm 进行全波长扫描,确定最大波长。结果表明:经过显色剂显色后,两者在417nm 处均具有稳定吸收,因此选择417nm为测试波长。
酸性染料的选择 取2mL 对照品溶液,在测试波长处,分别测定溴甲酚绿、溴百里香酚蓝2 种不同显色剂吸光度的大小,结果显示,在417nm 处,溴甲酚绿作为显色剂测得吸光度值相对较大,因此确定最佳显色剂为溴甲酚绿。
酸性染料用量的确定 取2mL 对照品溶液,分别置于不同的25mL 容量瓶中,加入配置好的显色剂溶液1.0、1.5、2、2.5、3mL,分别加入pH=5.0 的磷酸二氢钾缓冲液4mL 溶解,摇匀静置30min,加入氯仿5mL,震摇静置10min,取氯仿层,萃取两次,合并,测试波长下测吸光度,结果如图1 显示,随着酸性染料用量的增加,吸光度值逐渐递增,为保证结果准确,优选最佳用量为3mL。

图1 酸性染料用量试验数据
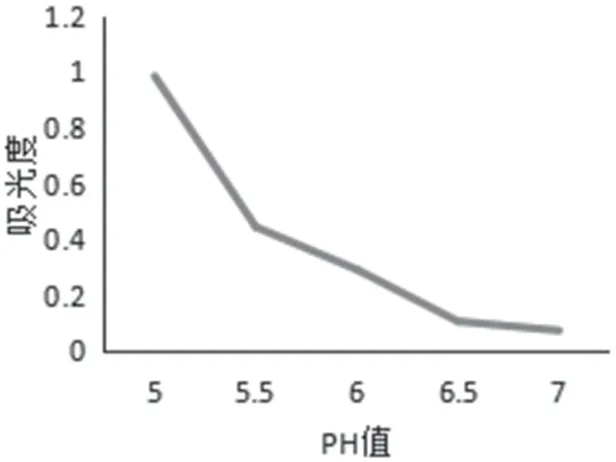
图2 缓冲液pH值试验数据
缓冲溶液pH 值 取2mL 对照品溶液,分别置于不同的25mL 容量瓶中,加入配置好的显色剂溶液,分别加入pH=5.0,5.5,6.0,6.5,7.0 的磷酸二氢钾缓冲液4mL 溶解,摇匀静置30min,加入氯仿5mL,震摇静置10min,取氯仿层,萃取两次,合并,测试波长下测吸光度,结果如图2 显示,随着PH 值的增加,吸光度值逐渐减小,由测定效果上来看,选出最佳pH 为5.0。
缓冲溶液用量的确定 取2mL 对照品溶液,分别置于不同的25mL 容量瓶中,加入配置好的显色剂溶液,分别加入pH=5.0 的磷酸二氢钾缓冲液2.5,3,3.5,4,4.5mL 溶解,摇匀静置30min,加入氯仿5mL,震摇静置10min,取氯仿层,萃取两次,合并,测试波长下测吸光度,结果如图3 显示,缓冲剂用量的吸光度值差别不大,且数值均较大,推测样品量过大影响了结果,但从细小的差距中,可以选出最佳pH 缓冲溶液用量为4.5mL。

图3 缓冲液用量试验数据
2.1.2 含量测定方法学考察 线性关系考察 精密吸取对照品溶液1、2、3、4、5mL,按照最佳显色方法测定,以甲醇作为对照,用紫外分光光度计在测试波长417nm 处测其吸光度,以对照品浓度(C)为横坐标,吸光值A 值为纵坐标,进行线性回归,结果见图4。

图4 线性关系
由图4,得出标准曲线为Y=0.0161X-0.0279,r=0.9992,在10.24~51.20µg·mL-1范围内盐酸小檗碱的线性关系良好。
精密度试验 精密吸取样品溶液2mL,显色后,测定吸光度。测定6 次。由试验结果得出其精密度RSD=0.144%,证明仪器精密度良好。
重复性试验 分别精密吸取样品溶液2mL,制备6 份供试品溶液,显色后测定吸光度。由数据RSD=2.66% 可得,其仪器重复性良好。
稳定性试验 精密吸取样品溶液2mL,分别在0,20,40,60,80,100,120,160,180 min,显色后测定其吸光度。结果显示,在3 h 内,吸光度值的平均值为0.3934,RSD=0.246%,因此可视仪器的稳定性良好。
加样回收试验 精密量取样品溶液2mL,共6份,每份分别加入对照品溶液2.5mL,作为供试品溶液,显色后测定吸光度。结果显示,平均回收率96.38%,RSD=2.61%,其加样回收试验良好。
2.1.3 提取方法考察 本试验研究考察超声提取、回流提取和渗漉提取3 种方法对总生物碱提取得率的影响。
超声提取法 称取药材细粉50.00g,量取12倍(600mL)75%乙醇,浸泡1h,在温度31℃,超声功率为100W 的条件下进行超声提取,提取2 次,每次1h,将两次超声提取的提取液合并,减压回收乙醇,在70℃水浴下浓缩的浸膏,将得到的浸膏放入真空干燥箱中干燥至恒重后,放冷后,进行称重。制备供试品,显色后测定吸光度。
回流提取法 称取药材细粉50.00g,量取12倍(600mL)75%乙醇,浸泡1h,进行回流提取2次,每次1h,两次提取的提取液合并,减压回收乙醇,在70℃水浴下浓缩的浸膏,将得到的浸膏放入真空干燥箱中干燥至恒重后,放冷后,进行称重。制备供试品,显色后测定吸光度。
渗漉提取法 称取药材粉末50.00g,量取适量75%乙醇,浸泡1d,进行充分的膨胀,将湿润膨胀后的药材,少量多次转移至自制的渗漉筒内,加入600mL 75%乙醇浸渍24~48h,以0.6mL·min-1速度进行渗漉,不断添加溶剂,保证溶剂高出药材,收集渗漉液,减压回收乙醇,在70℃水浴下浓缩的浸膏,将得到的浸膏放入真空干燥箱中干燥至恒重后,放冷后,进行称重。制备供试品,显色后测定吸光度。
供试品制备与测定 取提取物浸膏约0.20g,置100mL 具塞锥形瓶中,精密加入甲醇液50mL,称重,超声处理30min,放冷,称重,用甲醇液补足重量。过滤,精密量取续滤液1mL 于25mL 容量瓶中,加甲醇至刻度,摇匀,即得。以甲醇液为空白对照,于417nm 波长处测定A 值,从标准曲线上,计算出供试品溶液中盐酸小檗碱的浓度。结果表明对比3 种提取方法,渗滤提取药材中的生物碱最纯,但其提取的总量要小于回流提取,且时间最长,而超声提取虽方法简单易操作,但其提取的效率较其他两者不高。从提取总量上来看,提取方法优选回流提取法。见表1。

表1 考察试验结果
2.1.4 最佳工艺确定 正交试验设计 单因素试验考察中得出醇浓度在65%时,提取时间为1.0h,提取3 次时,料液比为1∶10,总生物碱提取量最好,根据醇浓度、料液比、时间、提取次数对总生物碱得率的影响,按照L9(34)表进行试验,各因素、水平见表2。

表2 正交表
正交试验 称取药材,共9 份,各50.00g,采用上述优先的最佳提取方法—回流提取,以提取物中总生物碱的含量为考察指标,优选提取工艺。按因素水平表设计的条件进行回流提取,提取液滤过,滤液合并,测定生物碱及小檗碱的含量,计算提取率。结果显示,在影响回流提取南天竹总生物碱的四个因素(醇浓度、提取时间、料液比、提取次数)中,根据极差值Ri 及方差分析,各因素对南天竹总生物碱提取率影响的主次顺序为D>C>B>A,即提取次数影响最大,其次为提取时间,可得出最佳的提取工艺为A2B1C2D3,即75%乙醇,1∶8 的料液比,提取3 次,每次1h。见表3。

表3 正交试验结果
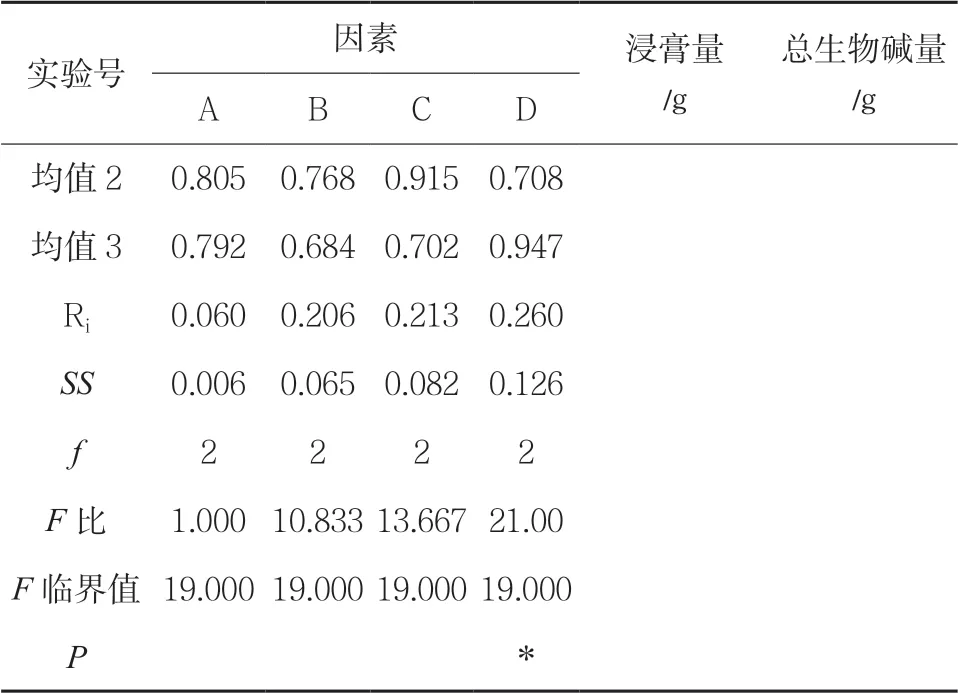
(续表)
2.1.5 验证试验 同一批药材称取3 份各50g,按照得到的最佳工艺条件提取,回收溶剂浓缩得浸膏,取浸膏0.2g 制备供试品,显色后测定吸光度,计算总生物碱量,实验平行3 次。如表4。结果,南天竹总生物碱的提取工艺稳定,得率平均为2.53%。

表4 验证试验结果
2.2 南天竹总生物碱纯化条件研究
2.2.1 树脂的选择及预处理 树脂的预处理 大孔树脂:用95%乙醇浸泡24h,充分溶胀,用乙醇洗至溶液加适量蒸馏水无白色浑浊现象时为止(取1mL 流出液加5mL 蒸馏水),最后用蒸馏水洗至无醇味,备用。阳离子树脂:称取适量阳离子树脂,用去离子水浸泡2h,去离子水洗至无色显中性,用2%HCl 冲洗至显酸性,再用去离子水洗至显中性,用2%NaOH 冲洗至显碱性(pH=8~9),将显碱性的阳离子树脂用去离子水洗至显中性,用2%HCl 冲洗至显酸性,再用去离子水洗至显中性,备用。
树脂的筛选 为较大程度的模拟生产过程,联系实际应用,本实验采用静态吸附以及动态吸附法选择树脂,分别用相同体积树脂以及相同质量的树脂进行比较。备选树脂分别为大孔树脂D101、ADS-17、NKA-9、NKA- Ⅱ、AB-8、HPD-722、阳离子树脂732。
2.2.2 上样溶液的制备 最佳提取工艺回流提取,汇总提取液,回收乙醇后,蒸干,去适量加水稀释,制得浓度为8mg·mL-1的生药液,备用。
2.2.3 静态吸附 称取不同型号大孔吸附树脂1.0 (湿),置100mL 烧杯中,加样品溶液20mL,每隔5min 振摇10s,持续2h,然后静置24h,充分吸附后,滤过,测定吸附前后溶液的吸光度值,计算静态吸附率。吸干经静态饱和吸附总生物碱后的各型号树脂表面水分,精密加入95%乙醇25mL,每隔5min 振摇10s,持续2h,测定洗脱液中总生物碱的质量浓度,计算饱和吸附量和解吸率。结果显示,ADS-17 的吸附率较低,D101、NKA-Ⅱ、AB-8、732 阳离子树脂的解析率较其他前两者低,从吸附率以及解析率来看,优选NKA-9 与HPD-722 两种进行动态实验。见表5。
饱和吸附量=(原料液质量浓度×原料液体积-流出液质量浓度×流出液体积) /湿树脂的量
吸附率=饱和吸附量/原料液中总碱的量× 100%
解吸率=洗脱液质量浓度×洗脱液体积/饱和吸附量×100%
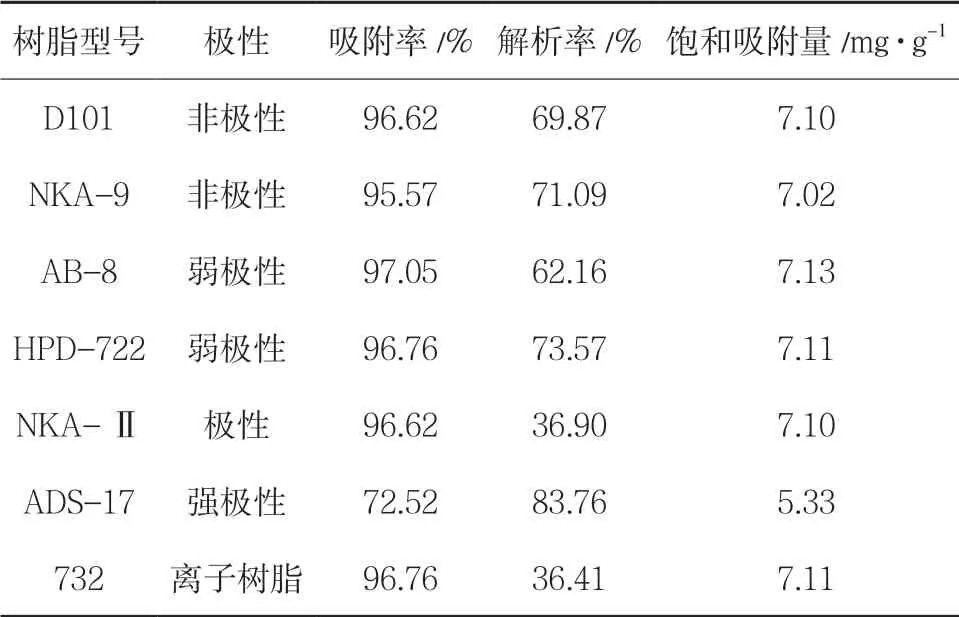
表5 静态吸附结果



表6 动态吸附结果
2.2.4 动态吸附实验 准确称取树脂适量,经预处理合格后湿法装柱,准确吸取上样液10mL,以流速0.5mL·min-1上柱,预吸附2h。先用9 倍蒸馏水以1.0mL·min-1流速洗脱,至洗脱液无色,再以80%乙醇0.5mL·min-1流速洗脱,洗脱至近无色,分别收集洗脱液,经浓缩干燥后得干燥物,计算各洗脱部分的总生物碱含量。结果显示:综合静态吸附实验及动态实验结果,HPD-722 比吸附量以及比洗脱量均略高于NKA-9,因此得出最佳树脂类型为HPD-722。见表6。2.2.5 动态吸附因素考察 上样液浓度考察 准确称取处理好的树脂3 份,每份树脂30g(湿重),浓度分别为8mg·mL-1(10mL),4mg·mL-1(20mL),2mg·mL-1(40mL) 的上柱液各 1 份,以0.5mL·min-1流速动态上样,然后用 6 倍蒸馏水以0.5mL·min-1流速洗脱,至洗脱液无色,再以 8 倍 80% 乙醇以0.5mL·min-1流速洗脱,至洗脱液无色,分别收集洗脱液。测定总生物碱含量,计算吸附率及洗脱率。结果显示:对比三者得到生物碱的纯度,得出浓度4mg·mL-1为最佳上样液浓度。
最大上样量 准确称取处理好的树脂30 g(湿重),取浓度为4mg·mL-1的上样液,以0.5mL·min-1的流速加入树脂中,用容器收集流出液,每10mL一管,测定收集液中的总生物碱的含量,绘制关于吸附的动态曲线。结果显示,当上样量的体积在20mL 左右的时候,生物碱类物质开始泄露增加,为减少损失,提高回收率,本实验选用最大上样量为35mL。结果见图5。

图5 动态吸附泄漏曲线

图6 动态洗脱浓度曲线
醇浓度考察 取处理好的树脂30g(湿重),制备样品溶液,吸取样品溶液20.0mL,以0.5mL·min-1流速动态上样,上样完毕后静置2h。然后用6 倍蒸馏水以0.5mL·min-1流速洗脱,至水洗脱液无色;再依次用 8 倍的35%、50% 、65%、80%、90%乙醇溶液进行洗脱,收集洗脱液。测定洗脱液总生物碱含量,计算不同浓度乙醇的洗脱率。结果显示,随着醇浓度的增加,洗脱率增加,醇浓度80% 以上时增加减少,洗脱效果接近,成本考虑,选择80%为最佳洗脱醇浓度。结果见图6。
洗脱用水体积 取处理好的树脂30g(湿重),吸取样品溶液,以0.5mL·min-1流速动态上样,静置2h,分段收集渗出液,计算渗出液中的总生物碱含量。结果显示,随着洗脱水体积的增加,浓度逐渐降低,从节约方面考虑,当水洗脱体积达到6 倍的时候,洗脱完全。因此最佳洗脱体积为6 倍。结果见图7。
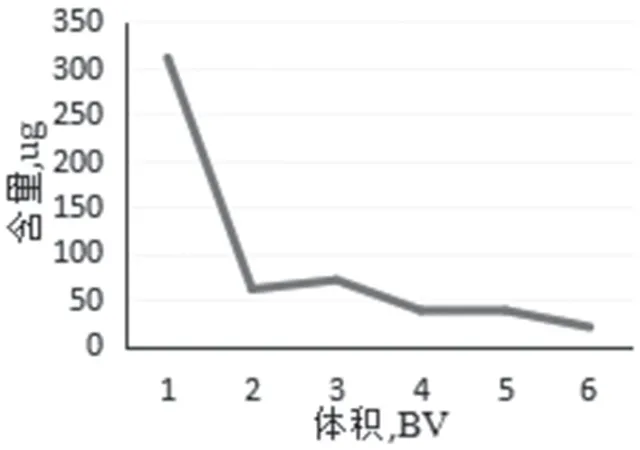
图7 动态洗脱用水体积曲线
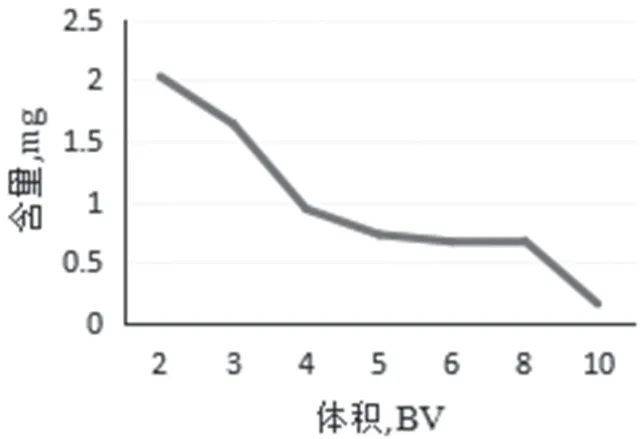
图8 动态洗脱醇体积曲线
醇洗脱量 取处理好的树脂30g(湿重),吸取样品溶液20.0mL,以0.5mL·min-1流速动态上样,上样后静置2h。然后用6 倍蒸馏水以0.5mL·min-1流速洗脱,再用80%乙醇以0.5mL·min-1流速进行洗脱,分段收集渗出液,计算洗脱液中总生物碱的含量。结果表明,随着体积的增加,浓度逐渐降低,对比总体及实验时间的考虑,当醇洗脱体积达到10 倍时,洗脱完全,因此,最佳洗脱体积为10 倍,结果见图8。
上样流速 取质量浓度为4mg·mL-1的4 份上样液20mL,缓缓加上,流速分别为0.5,1,2,3mL·min-1。上样完成后,静置2h,用6 倍蒸馏水洗脱,收集上样流出液和水洗脱液。测定总生物碱含量,计算吸附率。结果表明,随着流速的增大,吸附率开始下降,虽流速慢,吸附率大,但耗时较长,经综合考虑,选流速为2mL·min-1。
醇洗流速 按上述所确定的最优条件上样,先用6 倍水洗脱后,用 80% 乙醇 10 倍洗脱,流速分别为0.5,1,2mL·min-1,收集洗脱液。测定总生物碱含量。结果,随着流速的增加,洗脱率逐渐降低,醇洗流速0.5mL·min-1与1mL·min-1的结果差距不大,综合考虑时间及纯度比较,最佳醇洗流速为1mL·min-1。
2.2.6 验证试验 取处理好的树脂30g(湿重),按大孔树脂预处理下,湿法装柱并洗去杂质。制备样品溶液,吸取样品溶液15.0mL,以0.5mL·min-1流速动态上样,上样完毕后静置 2 h。然后按照得到的最佳纯化工艺进行纯化,收集洗脱液,测定总生物碱含量。实验平行3 次。如表7。结果,HPD722 大孔树脂纯化南天竹中总生物碱效果明显,总生物碱可从提取液中15.02%含量提升到70%以上。

表7 纯化验证试验结果
3 讨论
目前,对南天竹总生物碱的含量测定方法未见报道,南天竹生物碱主要是小檗碱类和原小檗碱类衍生物。文献检索发现,与上述母核结构相类似的总生物碱含量测定的方法多采用酸性染料比色法[7-8],该法主要针对生物碱类药物,样品用量少,灵敏度高,具有一定的专属性和准确度[9]。故本实验在文献的基础上,优先出南天竹总生物碱含量测定具体工艺。运用此方法需按照严格步骤操作,尽量减少人为误差。试验中所配制的酸性染料不宜使用时间过长,为保证数据偏差,建议在1 周内使用。生物碱于酸性染料形成的络合物易见光分解,注意避光。进行紫外测定时,浓度过大测量结果的误差增大,因此浓度不易过大。
本实验以小檗碱作为对照品,采用酸性染料比色,经正交试验得出,回流提取法为南天竹总生物碱的最佳提取方法,且最佳提取工艺为75%乙醇,1∶8 的料液比,提取3 次,每次1h。通过对影响大孔树脂吸附及解吸的各种因素系统研究,证明HPD-722 型树脂对于南天竹总生物碱具有吸附快、解吸较易、流体流动性能好、操作简单和周期短等优势,因而有利于规模化生产。经HPD-722 处理后的南天竹干浸膏中总生物碱的含量可达70% 以上。南天竹总生物碱含量明显提高。结果可为南天竹总生物碱相关制剂工艺奠定了良好基础,为大生产提供科学参考。
在进行树脂静态试验筛选时,树脂用量从节约样品以及实验时间上考虑,不易用量过多,建议与样品量比为1∶20。为保证测量准确,样品用量要高于树脂的吸附量。不论静态还是动态试验,要保证树脂有足够的时间进行充分吸附,建议静置24 h。随着吸附洗脱次数的增加,树脂内杂质增多,吸附力降低,考虑生产成本,保证总生物碱的回收率及纯度,建议使用3~4 次后必须对树脂进行再生处理。
