基于密度泛函理论的高岭石吸附机理研究进展
2020-02-25吕文婷张居兵卜昌盛郭若军王昕晔
吕文婷,程 运,谢 浩,2,张居兵,卜昌盛,郭若军,王昕晔
(1.南京师范大学能源与机械工程学院,南京 210042;2.南京师范大学镇江创新发展研究院,镇江 212000; 3.光大环保能源(镇江)有限公司,镇江 212004)
0 引 言
高岭土是中国的四大非金属矿之一(其余为云母、石英、碳酸钙),其主要组分是高岭石,以及埃洛石、水云母、伊利石、蒙脱石、石英和长石等矿物。高岭石属于1∶1型层状硅酸盐[1],层间通过氢键连接,层间距约为0.72 nm。高岭石晶体化学式为Al2O3·2SiO2·2H2O,晶体结构空间群为三斜P1,晶胞参数为a=0.51 nm,b=0.89 nm,c=1.45 nm,α=91.8°,β=104.5°,γ=90.0°,主要由Si-O四面体和A1-OH正八面体通过O原子连接组成[2-6],见图1。其中,Al原子均为六配位、Si原子均为4配位。

图1 高岭石的晶体结构Fig.1 Crystal structure of kaolinite
高岭土储量大且易于开采,产量大。2017年全球高岭土产量达到3700万吨,我国产量为320万吨[7-8]。高岭土用途广泛,主要用于制造纸张、陶瓷和耐火材料,其次作为涂料、橡胶填料、搪瓷釉料和白水泥原料使用,少量作为塑料、油漆、颜料、砂轮、铅笔、日用化妆品、肥皂、农药、医药等中的添加剂使用[7-11]。此外,高岭土比表面积较大,也可作为吸附剂使用。作为水体中的吸附剂使用时,吸附对象为水体悬浮物、油类有机物[12-14]和重金属离子[15-16]等;作为烟气中的高温吸附剂使用时,吸附对象为高温金属蒸气等[17]。前人主要通过试验手段研究高岭石吸附机理,但是难以深入理解吸附机制。近年来量子化学计算工具的普及促使越来越多的研究者从原子尺度分析高岭石吸附机理,以期在试验的基础上进一步理解吸附机制。
密度泛函理论(Density Functional Theory,缩写为DFT)是一种研究多电子体系电子结构的量子力学方法,在物理和化学上都有广泛的应用,是计算材料学和计算化学领域最常用的方法之一[18-23]。与其他解决量子力学的方法相比,基于密度泛函理论的计算结果较为令人满意。目前,已经有大量研究者将密度泛函理论用于高岭石吸附机理研究中,获得了很多重要结论[7,24-25]。
本文对近年来基于密度泛函理论开展的高岭石吸附机理研究进行了综述。首先,对密度泛函理论计算模型进行了介绍,具体包括:计算程序、计算步骤、吸附表面的构建以及泛函模型和计算参数的选取。随后,按照吸附物进行分类,从有机分子吸附、金属离子吸附、金属原子和分子吸附三个方面介绍了高岭石吸附的理论特征及其机理。最后,进行总结并提出了展望。
1 计算模型
1.1 计算程序
可进行基于密度泛函理论的量子化学/第一性原理计算程序包括:CASTEP(Cambridge Sequential Total Energy Package)、VASP(Vienna Ab-initio Simulation Package)、WIEN2k、PWSCF(Plane-Wave Self-Consistent Field)、DMol3、Gaussian等。
CASTEP基于平面波赝势理论,能且仅能计算周期性体系。其优点是,计算精度高,具有Windows和Linux两个版本[26-27]。Windows版本下图形化界面友好,适合初学者使用;Linux版本下计算效率更高且程序运行更稳定。VASP的前身是CASTEP1989版本,同样基于平面波赝势理论。其价格低于CASTEP,目前在材料模拟和计算物质科学研究中使用广泛,但其仅有Linux版本,初学者学习难度较大。PWSCF也采用了平面波赝势理论进行计算,也仅有Linux版本,但是计算效率低于VASP,其最主要的特点是免费且扩展性较好。WIEN2k使用平面波全电子势的方法,较VASP更为精确,且价格更为便宜,但是计算效率较低。除WIEN2k外,上述计算程序均在高岭石吸附计算中得到了应用。例如,Fu等[28]使用CASTEP研究了高岭石中掺杂过渡金属的结构和其电子性能,Kremleva等[29]使用VASP研究了铀酰在Al表面的吸附,Johnson等[14]使用PWSCF计算了有机分子(苯、正己烷、吡啶和2-丙醇)在高岭石表面的吸附。
DMol3是基于数值原子轨道基函数的计算程序,同时使用非局域化的分子内坐标,Hartree势多级展开,其特点是计算速度快,但是精度低于CASTEP。此外,DMol3不仅可以计算周期性结构,还可以计算非周期性结构,如团簇或分子等。例如,Lee等[20]使用DMol3计算了β-D-葡萄糖和纤维二糖在高岭石表面的吸附,Wu等[30]使用DMol3研究了H2O和Na(Ⅰ)对CO2在高岭石表面的吸附的影响。
Gaussian是一款综合性量子化学软件包,可以提供广泛的算法,其中包含了密度泛函理论的算法,通常用来计算分子反应且计算精度高。虽然Gaussian也具有周期性结构计算功能,但是计算效率非常差,因此极少应用于周期性结构计算。将周期性结构简化为团簇模型时,可以使用Gaussian进行计算。Wang等[21]使用Gaussian研究了2,4-二硝基甲苯在高岭石表面吸附,通过构造Si13O37H22四面体(Si-O)和Al6O24H30八面体(Al-O)的聚类模型以代替周期性的高岭石结构。
1.2 计算步骤
高岭石吸附计算步骤通常如下:首先,获得高岭石的晶胞参数,建立高岭石的晶体初始构型,对初始构型进行几何优化;接着,切割优化后的高岭石晶体构型,获得吸附表面,在表面上建造真空层;然后,构建超晶胞以扩大吸附表面,并再次进行几何优化;最后,将吸附分子、离子或原子放置在可能的表面吸附位置,对其进行几何构型优化计算,可以得到理论上最稳定即能量最低的吸附结构,见图2。通过布居分析、态密度分析、电荷密度分布和电荷密度差分等手段,可以得到吸附结构中原子间电荷转移和成键情况。通过过渡态计算可以得到表面的化学反应过程。此外,还可以计算获得红外光谱、X射线衍射光谱、核磁共振图谱等,与实验结果进行对比验证。
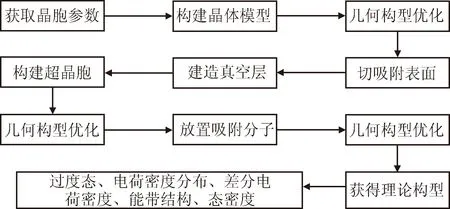
图2 吸附计算典型流程
Fig.2 Typical flow of adsorption calculation
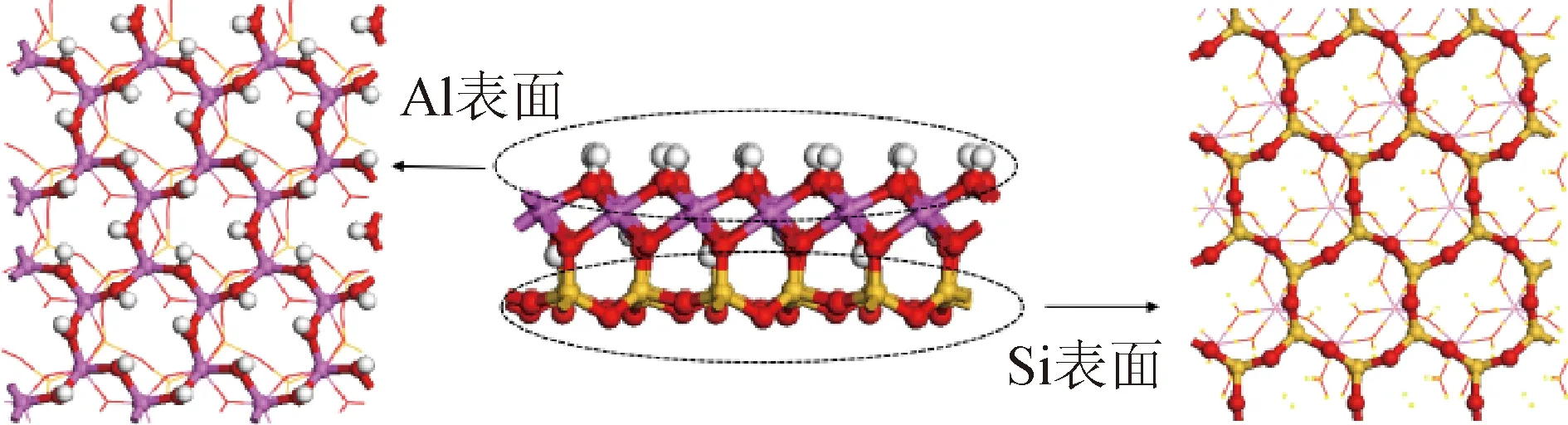
图3 高岭石Al和Si表面
Fig.3 Surfaces of kaolinite Al and Si
1.3 吸附表面
高岭石吸附研究通常针对Al表面和Si表面[31],见图3。表面构建的主要参数为真空层厚度和表面大小。真空层厚度通常选取1.0~6.0 nm,其大小主要由吸附分子的纵向长度决定,长度大则真空层厚度高,以避免吸附分子与上部的周期性晶胞下底面产生作用。Wang等[32]研究中的吸附物为Pb原子、PbO分子和PbCl2分子等(纵向长度为0.3~0.6 nm),所构建的真空层厚度为1.5 nm;Lage等[12]研究中的吸附物为沥青分子(纵向长度约为3.8 nm),所构建的真空层厚度达到5.6 nm。真空层厚度并非越大越好。真空层厚度越大,则计算体系越大,因此计算速度越慢。通常需要进行真空层测试以确定合适的真空层厚度。

表1 典型文献中使用的计算程序及计算参数Table 1 Calculation procedures and parameters used in typical literatures
Al表面或Si表面的大小通常选取2×1至3×3(1×1表示一个晶胞大小的表面),见表1和图4。其大小由吸附分子的大小决定,分子大则表面大,以避免周期性结构中吸附分子之间的相互作用。Wu等[30]研究中的吸附物为CO2(O-O距离为0.2 nm),高岭石吸附表面为2×2;Lee等[20]研究中的吸附物为β-D-葡萄糖(环状分子,最大对角宽度约为0.8 nm)和纤维二糖(双环连接状分子,长度约为1.2 nm),高岭石吸附表面为3×3。同样,表面并非越大越好。表面越大,虽然周期性结构中吸附分子相互间的作用更弱,吸附更接近于单分子吸附,但是计算体系越大,计算速度越慢。因为高岭土层与层之间通过氢键相连,属于弱连接,层与层之间的相互影响较小,所以大部分文献中采用单层高岭土模型进行吸附计算。

图4 高岭石表面构建示意图
Fig.4 Schematic diagram of kaolinite surface construction
1.4 泛函模型及计算参数
泛函包括LDA、GGA和B3LYP等方法,这些方法是为了处理体系中电子之间的相互作用。在CASTEP、VASP和DMol3中,常用的泛函模型LDA和GGA。但是LDA不能用于处理弱键体系,例如氢键。GGA方法中则有PW91,BP,PBE,BLYP,BOP,VWN-BP,PRBE,HCTH等泛函。高岭石吸附计算中,常用的模型组合为GGA-PW91和GGA-PBE,如Wang等[15]使用CASTEP中的GGA-PBE泛函计算Pb(Ⅱ)在高岭石表面的吸附;Wang等[19]使用了同样的计算程序和泛函来计算Cd原子、CdO、CdCl2分子在高岭石表面的吸附。也有学者使用LDA泛函来进行相关计算,如Zhao等[16]使用VASP中的LDA泛函计算Cd(Ⅱ)、Cu(Ⅱ)、Hg(Ⅱ)、Ni(Ⅱ)离子在高岭石表面的吸附;He等[33]使用了同样的计算程序和泛函来计算Pb(Ⅱ)离子在高岭石表面的吸附情况。
DMol3中的基组包括Min,DN,DND,DNP和TNP等,按照精度由低到高排列。其中Min精度最低,只用一个数值轨道基组;DN为双数值轨道基组;DND为非氢原子增加了d轨道函数极化;DNP为所有氢原子加入了p轨道函数极化,精度高,对氢键的计算更重要;TNP为所有的原子加入了极化函数,但目前只能使用H到Cl的元素(除He和Ne外)。高岭石吸附计算中一般选取DNP基组,如White等[18]的研究团队研究了高岭石高温脱羟基转变为偏高岭石过程中高岭石结构的变化,模拟时基组选择DNP;White等[34]结合DFT,以偏高岭石为例了解亚稳态原子结构,利用DMol3对偏高岭石进行优化,选取基组为DNP;Lee等[20]在研究β-D-葡萄糖和纤维二糖在高岭石表面的吸附时,泛函选取GGA-PBE,基组选择DNP。
最底层原子轨道中电子处理方法(Core treatment)包括:All electron,即把所有的电子都看成价电子;Effective core potentials(ECP),即用单个有效势代替内核电子来缩减计算成本;DFT Semi-core pseudopots (DSPP),即在计算中使用单个有效势代替内核电子,是特别针对DMol3模块开发的;All electron relativistic,即在处理所有电子的基础上对内核电子引入了相对论效应,计算时间最长。其中ECP和DSPP都是对21号以后的重元素进行处理,对于较重的元素,内核电子的速度接近光速,此时必须考虑相对论效应。高岭石吸附计算中,若吸附物中含有重金属元素,则可选取ECP方法或DSPP方法;若吸附物中只含有21号前的元素,则可选取All electron方法。
k点是专门针对周期性体系的参数。通常来说,k点取样越多,计算越精确,但是计算量越大,计算速度越慢。因此,需要进行k点测试以选取合适的k点数。高岭石吸附计算中,k点取值差异较大。
2 高岭石吸附的理论特征
2.1 有机物吸附
前人研究高岭土吸附有机分子的目的较为分散,例如:为了提升油砂提取沥青生产石油效率,需要掌握沥青组分在高岭土中的吸附机理[35-38];为了提高纸张质量,需要了解高岭石与纸张组分之间的作用原理;为了提升高岭土吸附有机污染物的性能,需要掌握吸附机理[39];为了增强高岭石疏水性能,需要了解有机分子在高岭石表面的吸附改性机理[40]。前人研究的有机分子包括:菲啶、苯并噻吩、四氢萘、萘、吲哚、苯、正己烷、吡啶、2-丙醇、β-D-葡萄糖、纤维二糖、2,4-二硝基甲苯、十二胺、油酸等。
有机分子在高岭石表面吸附时,通常是由于与Al表面形成的氢键。一般来说,其吸附的主要原因为有机分子中的N原子与表面上的H原子(轴向Al-OH)形成的氢键,以及有机分子中的H原子与表面上的O原子(轴向Al-OH)形成的氢键,和有机分子上的O原子与表面上的H原子形成的氢键,见图5。有机分子在Si表面上吸附的主要原因是,有机分子中的H原子与Si表面的O原子形成弱氢键,为了吸附更稳定,通常更倾向于吸附在Si环中心。例如,吡啶在Al表面吸附时,其中的N原子与Al表面上的H原子形成氢键而吸附,当吡啶垂直于表面时有利于氢键的形成;丙醇在Al表面吸附时,其中的O原子与Al表面上的H原子形成氢键而吸附[14]。

图5 有机分子与高岭石表面形成典型氢键
Fig.5 Typical hydrogen bonds formed between organic molecules and kaolinite surface
有机分子在疏水Si表面的吸附强度明显小于亲水Al表面[14],在Al表面的吸附能通常为10.0~50.0 kcal/mol,在Si表面的吸附能通常为0~10.0 kcal/mol。2,4-DNT在Al表面吸附时,分子中的O原子与Al表面上的OH形成氢键;Si表面吸附时,分子中的H原子与Si表面上的O原子形成了氢键。2,4-DNT优先吸附在Al表面的原因是与Al表面形成的氢键数大于与Si表面形成的氢键数[21]。
不同的吸附角度下,形成的氢键数量存在差别,通常来说,氢键越多,吸附越强。例如,萘吸附时与Al表面形成39°夹角,氢键数量最大;四氢萘吸附时与Al表面形成22°夹角,氢键数量最大[12];β-D-葡萄糖分子吸附时与表面形成约64.0°角时,形成的氢键数量最大[20]。
然而,氢键数量并非影响吸附角度的唯一因素,分子与表面的排斥作用也会影响。具有孤对电子的元素,如N原子等,与Si表面的O原子存在着排斥作用。例如,吡啶在Si表面吸附时,由于N原子与O原子存在排斥,N原子位于远离表面的一侧,吡啶中CH与Si表面O原子相互作用,吸附于Si环中心以增加CH-O的相互作用,所以吡啶垂直于表面吸附[14]。
有机物在水溶液中除了以分子形式存在,还可以离子形式存在。Liu等[40]研究了水溶液中十二胺和油酸及其极性头基团离子在高岭石表面的吸附。离子和分子吸附强弱没有固定顺序。十二胺的离子比分子吸附能强,油酸分子比离子吸附能更强。高岭石Al表面Ol周围呈电负性,平行羟基H原子周围呈电正性,Si表面呈电负性。十二胺阳离子可以同时吸附在Al表面平行羟基顶位和Si环中心穴位,油酸分子偏向于吸附在Al表面。这是由于吸附在高岭石表面后,十二胺极性头基团失去电子,油酸获得电子,但是油酸分子与离子吸附在Si表面时,与表面的静电作用几乎为零,吸附能非常小。
2.2 重金属离子吸附
水和土壤中的Pb、Cu、Hg、Cd、Ni和Zn等重金属离子容易在生物体中的富集,危害人类健康[41-42]。吸附法是最经济、最有效去除金属离子的方法之一,高岭石因其具有较好的吸附性能被应用于水体净化[43-48]。
一般情况下,重金属离子吸附在高岭石的Al表面。Al表面吸附位共有3种不同的类型,为顶位、桥位和穴位,见图6。不同重金属离子的稳定吸附位和表面吸附能差异较大,但是吸附都是由于离子与Al表面的O原子形成的共价键与离子键而产生。若重金属离子与水形成络合物,吸附是由于络合物水中的H原子与Al表面的O原子形成氢键而产生。前人试验研究发现,当吸附环境呈酸性时,重金属离子多吸附于Si表面,吸附位为边缘带电位和基底表面,见图7[49]。但是,目前没有该方面的DFT计算研究。

图6 重金属离子典型吸附位
Fig.6 Typical adsorption sites of heavy metal ions

图7 Si表面的两个吸附位点[49]Fig.7 Two adsorption sites on Si surface[49]
前人研究Pb(Ⅱ)离子吸附时,主要分为两种情况:一种是Pb(Ⅱ)离子吸附,另一种是Pb(Ⅱ)离子、PbOH(Ⅰ)离子、PbCl(Ⅰ)离子与水分子形成的配合物吸附。Pb(Ⅱ)离子吸附能范围在36.5~43.6 kcal/mol,Al表面的桥位吸附最为稳定[33]。吸附后,与Pb(Ⅱ)离子相邻的羟基由垂直或有一定角度变为与表面平行,Al表面上两个O原子向一个Pb(Ⅱ)离子提供电子,Pb(Ⅱ)离子与表面之间形成离子键。同时,O原子与Pb(Ⅱ)离子之间还存在共价键。随着吸附离子数量的增加,Pb(Ⅱ)离子覆盖范围增大,桥位和顶位吸附能也增大,Pb(Ⅱ)离子之间吸引力增大,促成形成Pb(Ⅱ)离子群。
Pb(Ⅱ)离子与水分子形成水配合物时,Pb(Ⅱ)离子配位数在1~8之间[50-51]。在Pb(Ⅱ)离子靠近Al表面时,由于高岭石的位阻效应以及Al表面羟基和水配体之间的排斥作用(平行于Al表面的羟基中的O原子,称Ol,对水配体中的H原子有很强的吸引,但垂直于Al表面的羟基中的O原子,称Ou,对配合水存在排斥作用),结合较弱的水分子会被挤走,所以吸附在表面的Pb(Ⅱ)离子配位数较Pb(Ⅱ)离子在水中的配位数低,为1~4。水配合物根据吸附位置不同,配位数和吸附能也不同,其范围在57.2~106.0 kcal/mol,大于单个Pb(Ⅱ)离子的吸附能。其原因是,Pb(Ⅱ)离子取代Al表面上的H原子与O原子成键的同时,水配体中的H也与表面上的O原子形成氢键,增强了吸附作用。Pb(Ⅱ)离子水配合物取代Al表面上的一个H而吸附时Pb(Ⅱ)离子倾向于吸附在与表面垂直的羟基(OuH)顶位,Pb(Ⅱ)离子水配合物取代Al表面上的两个H而吸附时Pb(Ⅱ)离子吸附可能性小,Pb(Ⅱ)离子水配合物取代Al表面上的三个H而吸附时Pb(Ⅱ)离子倾向于吸附在穴位[15],见图8。按水配合物中Pb(Ⅱ)离子取代表面H原子个数的不同,将其分别称为单齿配合物、双齿配合物和三齿配合物,不同配合水配体个数也有所不同。单齿配合时,Pb(Ⅱ)离子取代Al表面上的一个H原子与Ou原子成键,此时的Pb-O键与Al表面平行,水配体中H原子、O原子与Al表面O原子、H原子形成氢键。双齿配合时,Pb(Ⅱ)离子取代Al表面上的两个H原子与Ou原子成键,水配体与Al表面形成氢键,但高岭石空间的位阻效应增强,且OuH对水配体排斥作用增强,所以双齿配合物吸附能小于单齿配合物。三齿配合时,Pb(Ⅱ)离子取代Al表面上的三个H原子与Ou原子成键,水配体与Al表面形成氢键,此时Pb-O键在Al环中心成金字塔形,Pb(Ⅱ)离子吸附于Al环中心穴位。
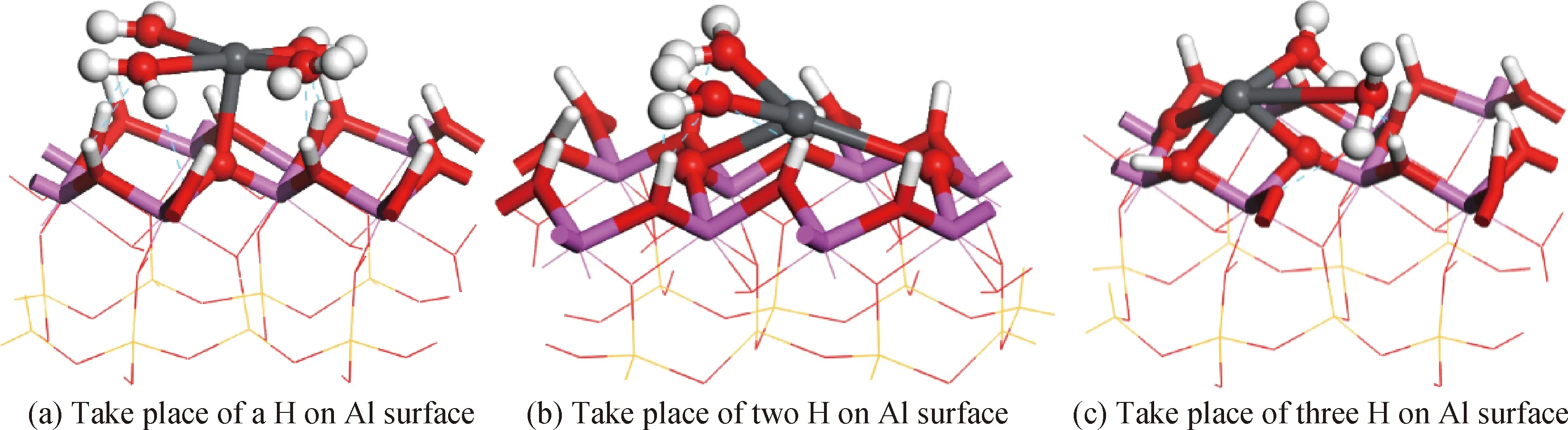
图8 Pb(Ⅱ)离子配合物的吸附
Fig.8 Complexes of Pb(Ⅱ) ion adsorption
PbOH(Ⅰ)离子、PbCl(Ⅰ)离子与水分子形成水配合物时,配位数均在1~5之间,吸附后配位数均为3~5[52-53]。吸附在高岭石表面时原理与Pb(Ⅱ)离子与水分子形成水配合物吸附一致,吸附能范围在55.2~182.6 kcal/mol和33.0~63.9 kcal/mol,单齿配合时,PbOH(Ⅰ)离子倾向于吸附在OuH顶位,双齿配合时,PbOH(Ⅰ)离子倾向于吸附在表面Ou-Ol桥位。
前人在研究Cu(Ⅱ)离子时,也分为离子的吸附和离子水配合物的吸附[16]。Cu(Ⅱ)离子偏向于吸附在顶部位点,随着Cu(Ⅱ)离子在Al表面的吸附密度增加,离子与Al表面上的H原子距离减小,吸附能增大,会产生吸附群的趋势。吸附密度(吸附的离子数量与吸附表面中原子数量的比值)从0.1增加到1.0时,Cu(Ⅱ)离子的吸附能从29.8 kcal/mol增加至34.4 kcal/mol。
Cu(Ⅱ)离子与水分子形成水配合物时,Cu(Ⅱ)离子配位数在1~6之间,吸附后配位数降低为2~3[54]。吸附在高岭石表面时原理与Pb(Ⅱ)离子与水分子形成水配合物吸附一致,吸附能范围在86.0~119.6 kcal/mol,单齿配合、双齿配合在表面的吸附位也与Pb(Ⅱ)离子一致。
前人在研究Zn(Ⅱ)离子时,主要为Zn(Ⅱ)离子与水分子形成的络合物吸附[55]。Zn(Ⅱ)离子与水分子形成水配合物时,Zn(Ⅱ)离子配位数在1~6之间,吸附后配位数为1~3。吸附在高岭石Al表面时原理与Pb(Ⅱ)离子与水分子形成水配合物吸附一致,吸附能范围在50.8~100.7 kcal/mol。在Al表面上,单齿配合时,Zn(Ⅱ)离子倾向于吸附在Ou顶位,双齿配合时,Zn(Ⅱ)离子偏向于吸附在Ou-Ou桥位。吸附在高岭石Si表面时,主要为Zn(Ⅱ)离子水配体中H原子与表面O原子形成氢键,吸附能为168.9 kcal/mol。
前人在研究其他重金属离子Ni(Ⅱ)、Hg(Ⅱ)、Cd(Ⅱ)离子等在高岭石表面吸附时,未考虑重金属与水分子形成络合物的情况,而仅分析了重金属离子吸附。Ni(Ⅱ)离子偏向于吸附在顶部位点,Hg(Ⅱ)离子偏向于吸附在穴位,Cd(Ⅱ)离子偏向于吸附在桥位。在上述位置的吸附能顺序为Ni(Ⅱ)>Pb(Ⅱ)>Cu(Ⅱ)>Cd(Ⅱ)>Hg(Ⅱ)[16]。Ni(Ⅱ)离子吸附时,电子从Ni(Ⅱ)离子向O原子转移,Ni(Ⅱ)离子与O原子之间同时存在离子键和共价键的成分,且键长是上述四个离子吸附中最短的,所以吸附能最强。Cd(Ⅱ)离子与O原子电负性不同,通过静电引力在桥位吸附,与两个O原子形成离子键。随着Cd(Ⅱ)离子,Hg(Ⅱ)离子在Al表面的吸附密度增加,也会产生吸附群的趋势,原理与Cu(Ⅱ)离子一致。但是,随着Ni(Ⅱ)离子覆盖范围增大,Ni(Ⅱ)-Ni(Ⅱ)离子间排斥力增强,离子与表面H原子距离增大,离子键和共价键减弱,因此吸附能减小[16]。吸附密度从0.1增加到1.0时,Cd(Ⅱ)离子吸附能从6.2 kcal/mol增加至11.3 kcal/mol,Hg(Ⅱ)离子吸附能从4.2 kcal/mol增加至9.2 kcal/mol,而Ni(Ⅱ)离子吸附能从59.6 kcal/mol降低至53.1 kcal/mol。
高岭石吸附水中的重金属离子的同时,也会吸附水分子。Johnson等[14]研究了水分子在高岭石表面的吸附情况。计算结果表明,水分子倾向于吸附在Al表面。水分子中的O原子与Al表面上的H原子形成氢键;水分子中的H原子与Si表面上的O原子存在弱氢键,为增强吸附,水分子吸附于Si环中心。水分子在Al表面吸附能为Si表面的约2倍。
2.3 金属蒸气吸附

图9 前人研究中的吸附Al表面Fig.9 Adsorption surface of Al in previous studies
固体燃料和生活垃圾中含有碱/重金属,燃烧时会产生不易捕集的亚微米颗粒物,排放至大气中会危害人类健康。高岭土在高温下具有重金属和碱金属蒸气吸附作用,掌握其吸附机理是提升其吸附性能的基础[56]。使用密度泛函理论计算研究重金属和碱金属高温吸附的文献报道较少,南京师范大学王昕晔等、东南大学黄亚继等以及华中科技大学姚洪等进行了相关研究[19,32,57-58]。重金属和碱金属在高温炉内(煤粉炉、垃圾焚烧炉)的主要形态为重金属氧化物蒸气、碱金属氢氧化物蒸气、金属氯化物蒸气以及少量的金属原子蒸气等。前人主要针对Pb、Cd、Na和K的上述形态进行了计算研究[19,32,57-59]。与有机分子吸附和重金属离子吸附研究不同的是,重金属蒸气吸附发生在700 ℃以上的高温条件下,高岭土表面羟基会发生脱落,向偏高岭土转变[60-62]。因此,吸附表面并非高岭石原表面。前人对不同的吸附表面进行了研究,主要为脱去一个H2O分子(-OH+-OH → -O+H2O)的Al表面、完全脱羟基Al表面和Si表面,见图9。
金属原子在高岭石表面的吸附主要是金属原子与表面O原子成键,其中Al表面需要脱除羟基才可以暴露出O原子,发生吸附,见图10。重金属原子无法吸附在Si表面,碱金属原子则可以吸附。在Si表面,Pb原子不与表面成键,Cd原子与表面存在排斥力。Na原子和K原子偏向于吸附在Si环中心穴位,属于化学吸附。在脱除一个羟基的Al表面(标记为Al-1),Pb原子吸附在失去羟基的两个五配位Al原子之间,其中Pb原子与附近的Al原子和O原子均存在相互作用,同时具有共价键和离子键特征,属于化学吸附;Cd原子吸附在顶位,而仅与O原子存在较弱的相互作用,属于物理吸附。在完全脱羟基表面(标记为Al-2),Pb原子和Cd原子与表面的作用增强,吸附能均为Al-1表面的两倍,其中Cd原子吸附仍然为物理吸附,Pb原子和Cd原子吸附位置均为中心穴位偏向表面暴露的四配位Al原子的位置。Na和K原子吸附在Al环中心穴位而无偏移,Na原子与表面上的O原子形成了离子键,K原子与表面O原子作用较弱,均为化学吸附。
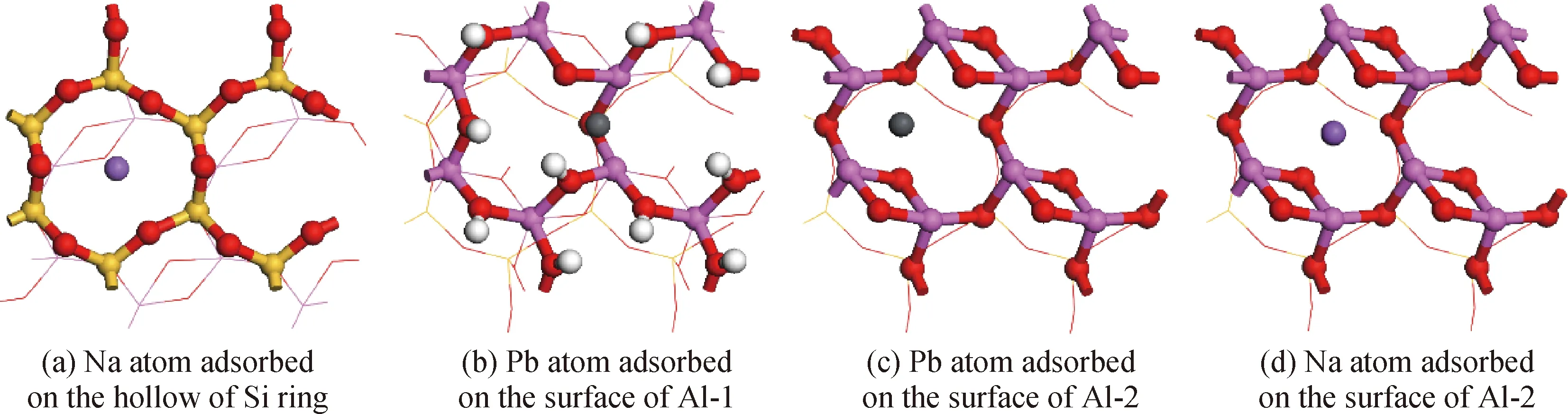
图10 金属原子典型吸附位
Fig.10 Typical adsorption sites of metal atoms
重金属氧化物分子和碱金属氢氧化物分子在高岭石表面的吸附主要是氧化物/氢氧化物中的O原子与表面Al或Si成键,同时金属原子与表面O原子成键,见图11。在Si表面,PbO分子和CdO分子与表面基本无作用,因此无法吸附;NaOH分子和KOH分子中的-OH与Si原子则可以发生强作用,导致Na原子和K原子与-OH断裂,碱金属原子吸附于Si表面的中心穴位、-OH成为Si表面的羟基。在Al-1表面,PbO分子吸附在Al环脱羟基的缺口处,PbO分子中的O原子与表面上的两个五配位Al原子成离子键、Pb原子与表面上的两个O原子成共价键;CdO吸附原理与PbO一致,二者吸附能相近。在Al-2表面,PbO,CdO与表面同时形成共价键和离子键,且吸附能为Al-1表面的两倍;NaOH分子中的-OH与表面两个四配位Al原子作用,成为其羟基,Na原子位于Al环中心穴位,与Si表面吸附不同的是,Na与-OH并未发生断裂,KOH分子吸附与NaOH分子吸附类似。碱金属氢氧化物分子和重金属氧化物分子在Al-2表面的吸附能相近,而其原子吸附能差异较大,因此认为,氧化物/氢氧化物吸附作用主要来源于吸附物中的O原子与表面不饱和Al原子的作用。

图11 金属氧化物典型吸附位
Fig.11 Typical adsorption sites of metal oxides
金属氯化物分子在高岭石Si表面的吸附作用来源于金属原子与Si表面的作用,而Cl原子与表面存在排斥作用,见图12。因为重金属原子与Si表面无作用,所以重金属氯化物分子无法吸附在Si表面。而碱金属原子与Si表面存在吸附作用,所以NaCl分子和KCl分子分别以化学吸附和物理吸附的形式吸附在Si表面。金属氯化物在Al表面的吸附作用与表面是否脱羟基密切相关。在仅脱除一个H2O的Al-1表面,PbCl2分子和CdCl2分子中的重金属原子分别吸附于O原子上方的顶位和两个O原子间的桥位,Cl原子与表面羟基形成氢键。高温作用下,Cl原子可以与羟基中的H原子作用,生成HCl,最终重金属原子与羟基残留O原子形成氧化物分子形式,稳定的吸附在Al表面。对于彻底脱羟基的Al-2表面,Cl原子与四配位Al原子产生强作用,生成共价键。碱金属氯化物分子吸附与重金属氯化物分子吸附形式类似,但吸附能高于重金属氯化物分子吸附。

图12 金属氯化物典型吸附位
Fig.12 Typical adsorption sites of metal chloride
3 结 语
基于密度泛函理论的高岭石吸附计算主要使用CASTEP、VASP、DMol3等计算程序,吸附表面通常包括Al表面和Si表面,表面大小通常为2×1~3×3,真空层厚度通常为1.0~6.0 nm,具体尺寸视吸附物尺寸和吸附姿态而定。前人的研究主要集中在高岭石吸附有机分子、重金属离子和金属蒸气等方面。
高岭石吸附有机物分子的主要原因是氢键的形成,包括:有机分子中的N原子与Al表面上的H原子形成的氢键、有机分子中的O原子与Al表面上的H原子形成的氢键、有机分子中的H原子与Si/Al表面上的O原子形成的氢键,形成的氢键越多,吸附能越大。有机分子在Si表面的吸附能明显小于Al表面。
高岭石吸附重金属离子的主要原因是离子与Al表面的O原子间成键,Al表面上存在三个吸附位点:顶位、桥位和穴位。Pb(Ⅱ)离、Cu(Ⅱ)离子吸附在Al表面存在两种形式,一是Pb(Ⅱ)离子、Cu(Ⅱ)离子直接吸附,另一种是Pb(Ⅱ)离子、PbOH(Ⅰ)离子、PbCl(Ⅰ)离子和Cu(Ⅱ)离子与水形成络合物后吸附,后者具有更大的吸附能。对于其他重金属离子如Ni(Ⅱ)、Cd(Ⅱ)、Hg(Ⅱ)等,吸附位存在差异,且吸附能受吸附密度影响。
高岭石高温吸附重金属/碱金属蒸气时会发生高温脱羟基,因此存在部分脱羟基和全部脱羟基两种情况。重金属蒸气分子仅能吸附在Al表面而碱金属分子可以吸附在Al表面和Si表面。不同的金属蒸气分子形态(原子态、氧化物分子、氢氧化物分子、氯化物分子)在表面的吸附机理不同。总体上看,脱羟基后的不饱和Al原子是重金属/碱金属蒸气吸附的主要原因。
尽管理论计算已经为高岭石吸附提供了很多机理性的结论,但是相关的研究还不够充分,还有较多值得进一步计算研究的内容,具体如下:
(1)研究Ni(Ⅱ)、Cd(Ⅱ)和Hg(Ⅱ)等重金属离子在高岭石表面吸附时,进一步考虑其与水分子形成的络合物的作用,同时考虑其与Cl-、OH-等离子形成的一价离子与水分子形成的配合物,并且考虑Si表面的吸附,从而进一步阐述重金属离子在高岭石表面的吸附机理。
(2)除了Pb、Cd、Na、K外,还需要对其他重金属原子/分子(Cr、Co、Fe、As等)在高岭石表面的吸附机理进行研究,在理论上拓展高岭土可能的吸附应用范围。
(3)前人研究较多针对无缺陷的高岭石表面,而缺乏对高岭石表面缺陷对吸附影响的认识。例如,高岭石表面羟基脱落缺陷、高岭石表面Si原子或Al原子脱落缺陷等。对于这些缺陷的认识将对高岭土改性具有重要指导意义。
