HCV 3b亚型全长序列测定及基线耐药相关置换的分析
2020-02-08黄杰庭单振刚尤庆柱付涌水
黄杰庭,许 茹,廖 峭,王 敏,单振刚,尤庆柱,戎 霞,4,付涌水,4
1 广州血液中心,广州 510095; 2 广州市医学重点实验室(血液安全重点实验室),广州 510095;3 韶关市第一人民医院 耳鼻喉科,广东 韶关 512000; 4 南方医科大学 检验与生物技术学院输血医学系,广州 510515
我国是HCV高流行地区,约有1000万感染者[1],其中75%~85%的感染者发展为慢性丙型肝炎(CHC),后者有可能进一步发展为肝纤维化、肝硬化、甚至肝癌或肝衰竭等[1-2]。HCV感染已经成为我国不容忽视的公共卫生问题。
HCV具有高度异质性,根据基因序列的差异程度可分为7种基因型和超过80种亚型,HCV准确分型的金标准是基于病毒全长序列的系统进化分析[3]。目前主要通过分区段扩增和一代测序,把各段序列进行拼接得到HCV全长序列[4]。然而,由于HCV基因组的高度变异,这种方法不易获得病毒全长序列,而且分区段扩增的策略增加了实验操作的工作量和工作难度。此外,一代测序的灵敏度较低,仅能检测丰度超过15%的突变或变异。因此,建立一种快速且高灵敏度的HCV全长序列测定方法,仍是当前HCV研究热点之一。
近年来,多种针对HCV的直接抗病毒药物(direct-acting antivirals, DAA)已进入临床应用[5]。我国从2016年4月开始陆续引进和批准sofosbuvir等6种DAAs上市[6]。此前的标准治疗方案为聚乙二醇干扰素联合ribavirin,与其相比,DAA治疗方案具有更高的持续病毒学应答(sustained virological response, SVR)率、疗程短和不良反应少等优点[5]。然而,仍有部分感染者未获得SVR或出现停药后复发,与基线耐药相关置换(resistance associated substitution, RAS)密切相关[7]。命名RAS需要确定对应的HCV蛋白以及氨基酸位点。以NS3 Q80K为例,RAS的首字母(Q)为野生型氨基酸,然后是氨基酸的位置(NS3蛋白的第80位),后面跟的字母表示耐药突变的氨基酸(K)[7]。体外研究[7]表明,NS3 Q80K可使simeprevir的抗病毒作用降低10倍;临床使用daclatasvir联合sofosbuvir治疗HCV基因3型感染者时,无NS5A基线RAS患者的12周SVR率高达92%,而存在NS5A基线RAS患者则仅有54%。因此,对HCV感染者在进行个体化DAA治疗前检测基线RAS以选择最佳药物组合和治疗方案,尤为重要。
本课题组在前期研究中通过基因片段扩增,发现1例献血者可能感染了HCV 3b亚型。该亚型在广州地区十分少见。在Genbank数据库中,HCV 3b亚型的全长序列仅有13条,其中分离自中国的仅有2条。研究[8]发现感染HCV 3型(主要为3a亚型)的患者中,NS5A Y93H和A30K两个基线RAS的频率均为5%~10%。本研究采用二代测序法(next generation sequencing,NGS)分析该献血者的HCV全长序列和基线RAS,为HCV 3b在广州地区的分子流行病学研究和DAA治疗方案的制订提供科学依据。
1 材料和方法
1.1 研究对象 实验编号为2584的无偿献血者(男性,30岁)于2016年7月在广州血液中心参与无偿献血,血液筛查显示其抗-HCV和HCV RNA呈反应性,无HBV、HIV和梅毒螺旋体感染,ALT水平升高(203 IU/ml)。1个月后对该献血者随访,抽取静脉血5 ml,EDTA抗凝,离心后将血浆置-20 ℃保存,用于HCV RNA定量和NGS。本研究方案经由广州血液中心医学伦理委员会审批(编号:广州血液中心医伦〔2019〕第40号),研究对象签署知情同意书。
1.2 HCV RNA定量 研究对象的随访血浆标本送至广州金域医学检验中心进行HCV RNA定量,采用实时荧光定量PCR法(Roche COBAS AmpliPrep/COBAS TaqMan,德国罗氏诊断有限公司)。
1.3 血浆病毒核酸的提取 采用Ng等[9]报道的方法,将血浆标本经0.45 μm滤器过滤去除细胞碎片后,取120 μl加入核酸酶混合物(RNase One Ribonuclease,普洛麦格生物技术有限公司;Turbo DNase,赛默飞世尔科技有限公司)于37 ℃孵育1.5 h以去除游离核酸。随后使用QIAamp viral RNA mini kit(凯杰生物工程有限公司),按照说明书的步骤提取病毒RNA。
1.4 序列非依赖性扩增 采用Ng等[9]报道的方法,以带有9个随机碱基(N)和18个固定碱基的逆转录引物(GCCGACTAATGCGTAGTCNNNNNNNNN),采用SuperScript Ⅲ First-Strand Synthesis Kit(赛默飞世尔科技有限公司)将病毒RNA逆转录为cDNA。然后采用Platinum Taq Hot-Start PCR Master Mix(赛默飞世尔科技有限公司)对cDNA进行PCR扩增,PCR引物序列与逆转录引物的18个固定碱基一致。
1.5 二代测序(NGS) 上述PCR产物送至上海派森诺生物科技有限公司,采用Illuminate Hiseq进行2×150 bp双末端测序。
1.6 测序数据的处理分析 参照Thomson等[10]报道的方法,对NGS获得的原始序列(Raw reads),采用Babraham Bioinformatics(https://www.bioinformatics.babraham.ac.uk)的FastQC和Trim_galore分别去除低质量序列和引物/接头序列,得到高质量序列(HQ reads)。后者经Bowtie程序(http://bowtie-bio.sourceforge.net/index.shtml)处理去除人类基因组序列,余下的序列通过Tanoti(http://bioinformatics.cvr.ac.uk/tanoti.php)与187条HCV全长参考序列比对得到HCV reads。最后将HCV reads通过Tanoti的从头组装(de novo assembly)拼接得到HCV全长序列。该序列已上传至Genbank(https://www.ncbi.nlm.nih.gov/genbank/),进入号为MN385583。通过HCV-GLUE(http://hcv.glue.cvr.ac.uk/#/aboutGlueProject)进行RAS分析[10]。
1.7 HCV基因分型 采用本课题组此前建立的方法[4],通过MEGA7.0,将2584号标本的序列和HCV各基因型/亚型参考序列构建系统发生树,获得准确基因分型。
2 结果
2.1 NGS测序结果 2584号标本的HCV RNA定量为1.57×107IU/ml。NGS共获得8.4 Gb数据,raw reads数为56 092 306。去除引物序列、接头序列以及低质量序列后,高质量序列(HQ)数为40 694 590,占72.5%。HCV reads数为33 295 509,占HQ reads的81.8%。
2.2 HCV全长序列分析 如图1所示,2584号标本的HCV reads拼接后得到的序列对HCV基因组的覆盖率为100%,即获得病毒全长序列,平均测序深度为488 007。
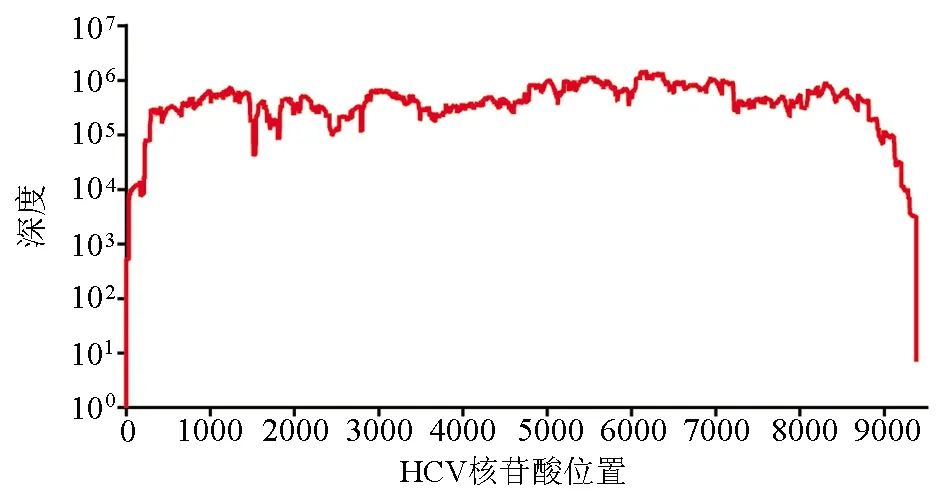
图1 2584号标本HCV核苷酸位点的测序深度
2.3 病毒基因分型 通过MEGA7.0对2584号标本的病毒全长序列和参考序列构建系统发生树(图2)。HCV基因1型-7型(gt1-gt7)形成7个不同的簇。2584号标本(红点)位于HCV gt3的簇上,与13株3b亚型同源性最高,形成独立分支,表明此标本属于3b亚型。从3b的分支(图2右)可见2584号标本和另外2株分离自中国的序列均位于C簇上,明显独立于A簇和B簇,提示我国的HCV 3b可能具有独特的分子流行病学特征。
2.4 RAS分析 2854号标本的NS3、NS5A和NS5B RAS如表1所示。其中NS5A Q/A30K和L31M的丰度分别为99.16%和98.37%,属于高丰度RAS。此外,还发现了10个低丰度RAS(<0.5%),包括NS3 Y56H、Q80K、Q80R和A156G,NS5A M28G、Q/A30G、L31F和Y93H,以及NS5B S282T和V321A。
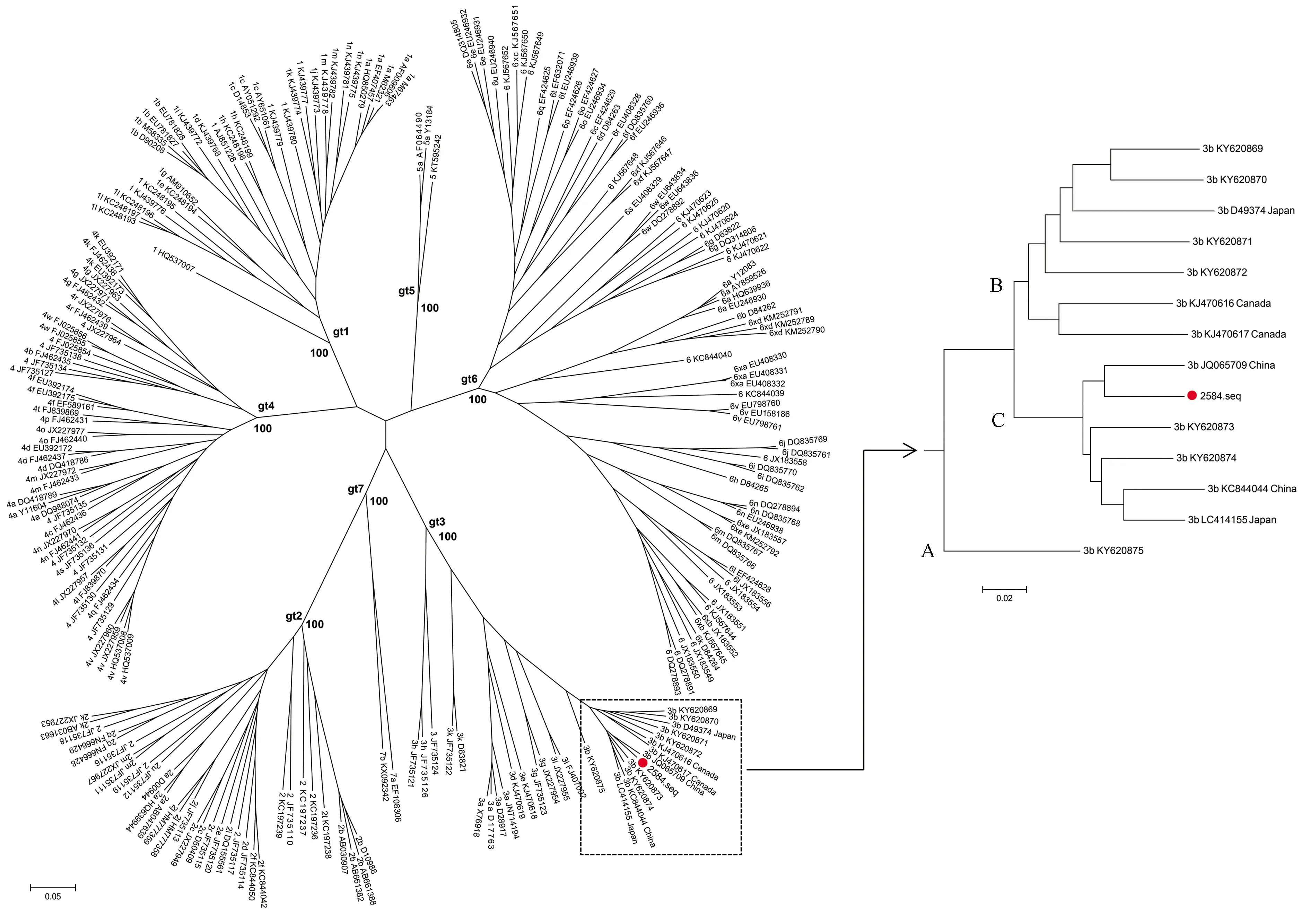
注:虚线方框为3b亚型,红点标示为2584号标本。

表1 2584号标本的RAS分析
3 讨论
HCV的基因型/亚型与病毒的传播途径,感染的发生、发展和治疗方案密切相关,而且在不同地域的分布存在明显差异。本研究的2584号标本属于3b亚型,在广州则较为少见,仅占HCV慢性感染者的5%[11]。受分段扩增和一代测序的限制,HCV 3b的全长序列很难获得,迄今我国仅有2条3b全长序列上传到Genbank。本研究采用NGS和生物信息学分析方法,成功获得2584号标本的全长序列。与基于一代测序的传统方法相比,本方法具有以下优点:(1)采用序列非依赖性扩增,避免了特异性引物在扩增时引入偏倚;(2)大大减少了实验操作的工作量和工作难度;(3)超大的数据量;(4)更高的灵敏度以检测低丰度变异。
DAA因具有高SVR水平、不良反应发生率低以及缩短治疗时间等优势,已被认为是治疗CHC的首选药物[12]。根据药物作用靶点的不同,DAA分为NS3/4A蛋白酶抑制剂、NS5A抑制剂和NS5B聚合酶抑制剂[5]。然而亦有DAA治疗无效的病例报告,经研究发现与RAS密切相关。这些RAS发生在患者所用DAA的靶基因上,导致药物敏感性下降甚至无效[7]。通过临床证据和体外试验,目前已鉴定出约50个RAS[12]。HCV不同基因型/亚型之间的RAS和基因耐药屏障不尽相同[7]。基线RAS影响着临床治疗方案的确定,包括DAA的选择和疗程的长短等。欧洲肝病学会建议,采用DAA治疗CHC患者前需进行RAS检测,尤其是选择NS5A抑制剂前应检测NS5A 24~93位氨基酸序列是否含有基线RAS[12]。本研究在2584号标本发现NS5A Q/A30K和L31M两个高丰度RAS,可能会导致对daclatasvir等6种DAAs无效。笔者也分析了Genbank的13条HCV 3b的全长氨基酸序列,发现其中12条也携带Q/A30K和L31M,1条携带Q/A30K,提示携带两个基线RAS的3b亚型病毒株出现的频率可能相当高。值得注意的是,虽然这两个RAS导致3b亚型对NS5A抑制剂耐药已在体外试验中被证实,但未见相关临床报道。这可能与3b亚型少见于欧美国家有关。此外,虽然一般认为RAS的丰度超过15%才会影响DAA的疗效,但亦有研究[13-14]指出,低丰度RAS在DAA的选择压力下可成为优势毒株导致治疗无效或复发。本实验在2584号标本检测到10个低丰度RAS(<0.5%)。由于有着足够的测序深度,这些RAS的reads均>25,表明不是测序错误,而是病毒序列中真实存在的变异。后续研究拟对该献血者进行随访,尤其是DAA治疗后的病毒核酸定期监测,以了解低丰度RAS是否影响该献血者的DAA疗效。
本研究建立的基于NGS的HCV全长序列测定和RAS分析方法可进行进一步优化。对于病毒载量高的标本,后续试验在保证序列拼接质量的前提下,可适当降低NGS数据量和测序深度,以获得更好的成本效益。此外,近年来,以单分子测序为技术核心的第三代测序技术(third generation sequencing, TGS)迅速发展。TGS最主要的优势在于超长的读长,可达30 kb甚至超过100 kb,远远超过NGS的100~400 bp[15]。超长的读长使得序列拼接更准确,也使得病毒准种的研究成为可能。然而,就当前技术水平而言,TGS尚未能广泛应用于HCV或其他研究,主要是因为TGS在文库构建和测序的过程中没有核酸扩增的步骤,因而对待测样本的核酸浓度要求相当高。从血浆中提取的病毒核酸,浓度往往很难达到TGS建库要求。此外,TGS的测序错误率较高,不适用于测定基因组变异程度很高的HCV序列。总之,NGS是目前测定HCV全长序列的最好方法,但随着技术水平的不断发展,TGS有着更广阔的应用前景。
