利用高通量测序快速定位目标性状位点的研究
2020-01-07赵青松张孟臣杨春燕赵团结
林 静,赵青松,张孟臣,杨春燕,赵团结
(1.河北省农林科学院 粮油作物研究所,国家大豆改良中心石家庄分中心,农业部黄淮海大豆生物学与遗传育种重点实验室, 河北省农作物遗传育种重点实验室,河北 石家庄 050035;2.南京农业大学国家大豆改良中心,农业部大豆生物学与遗传育种 重点实验室(综合),国家作物遗传与种质资源重点实验室,江苏 南京 210095)
大豆(GlycinemaxL.)是我国主要的粮、油、饲兼用作物。随着我国经济水平发展和人民生活水平的日益提高,我国对大豆的需求总量与日俱增。据报道,我国近几年大豆消费量维持在1亿t以上,而我国大豆年生产量仅为1 200万t左右(http://www.stats.gov.cn/),自给率不足20%,大豆严重依赖进口的现状对我国粮食安全造成巨大的隐患[1-3]。目前,我国大豆单产水平在1.8 t/hm2左右,而大豆前三大生产国美国、阿根廷和巴西的单产水平分别为3.51,3.02,2.91 t/hm2[4],因此,我国大豆单产有很大的提升空间,通过提高我国大豆单产水平,可以一定程度上缓解对进口大豆的依赖性。
常规的作物育种需要通过杂交、选育、株行观察、加代稳定、产量鉴定、产量比较、区试和生产试验等步骤[5],整个育种周期需要5~10 a。常规品种选育一般至少需要5代以上,要求主要农艺性状稳定。然而,在实际选育过程中,即使到F5以上,选育株行中仍会出现农艺性状分离,其对育种家造成很大的困扰。随着分子技术的发展,分子辅助育种(Marker assistance selection, MAS)被应用到现代育种当中,利用与性状关联的分子标记可以快速筛选出有利于生产的纯合性状[6],不仅减轻了田间工作量,并且显著缩短了育种周期。因此,鉴定与目标性状关联的分子标记,即定位调控目标性状的关键位点是分子辅助育种的基础。
目前,常用基因定位的遗传群体有F2、RIL、DHL和自然群体等。其中F2群体为临时群体,一般用于定位质量性状位点(Qualitative trait locus)效果较好[7-9], RIL和DHL则为稳定遗传的永久性群体,适用于需要多重复的数量性状位点(Quantitative trait locus, QTL)的定位[10-11]。自然群体属于非人工构建的,经过自然驯化而成的群体,其优点是含有丰富的遗传多样性,而缺点则是不适用于鉴定微效的QTL位点[12-13]。另外,自然群体往往受群体大小的限制,多种情况下仍然难以获得调控目标性状关键基因的精确位置。因此图位克隆(Map-based cloning)往往需要扩大遗传群体。而数量性状由于受遗传背景的影响,需衍生出次级群体,例如剩余杂合系(Residual hybrid lines, RHL)和近等基因系(Near-isogenic line, NIL)等,进而进行精细定位和图位克隆,目前RHL和NIL已经广泛用于水稻、玉米、小麦和大豆等作物的精细定位研究[14-17]。
图位克隆从方法和策略上已日趋成熟,但构建基本遗传群体、鉴定性状和构建次级群体,过程繁琐且耗时耗力。尤其是次级群体RHL和NIL的构建往往需要连续回交或自交至4~5代以上,才可以将90%以上的遗传干扰去除。而育种过程中,F5以上的单株行如发生性状分离,其本身就包含RHL和NIL 2种特性,但由于检测技术手段的限制往往被忽略。基于以上研究现状,本研究利用高世代的中间材料,以调控大豆花色的基因位点W1为例,在田间F6株行中筛选花色分离的家系,运用近等基因系原理和重测序技术快速挖掘调控目标性状的遗传位点,进而阐释本策略的有效性,结果将提供一种可以快速挖掘调控目标性状遗传位点的定位策略,以加速育种进程。
1 材料和方法
1.1 试验材料及性状鉴定
试验材料选用大豆MS轮回群体中364个F6株行,于2017年在河北石家庄堤上实验站进行种植,行长6 m,每行播种50~100粒,在开花期对株行的花色进行统计观察,对出现分离的株行进行单株收获形成F6∶7。分别取F6∶7每个家系的种子20~30粒,于2018年继续进行株行观察花色分离情况。
1.2 DNA样品采集与重测序方法
2017年对出现分离的株行单株进行挂牌,每单株取1~2片嫩叶冻存备用。2018年对F6∶7每家系进行观察,统计分离情况。在每个不发生分离的株行中随机挂牌1株,取1~2片嫩叶,并将株行表型一致的叶片混合冻存备用。
重测序在广州基迪奥生物科技有限公司进行。具体方法如下:利用商用基因组试剂盒提取植株DNA。运用NEB建库试剂盒(NEBNext® ΜLtraTMDNA Library Prep Kit for Illumina®)构建测序文库。测序均在Illumina测序平台进行。将去除接头后的测序数据和参考基因组Wm82.a2.v1进行比对,参数设置为“mem 4-k 32-M”[18],SNP变异运用GATK′s Unified Genotyper软件进行分析。
1.3 dCAPS标记开发和检测方法
在重测序结果基础上,利用鉴定到的SNP变异开发目标区间的dCAPs标记。具体流程方法如下:明确在目标区段的SNP变异位点,在Phytozome v.12(https://phytozome.jgi.doe.gov/pz/portal.html)下载SNP变异位点上下游各30 bp的DNA序列,将野生型和突变类型的DNA序列同时输入至网站http://helix.wustl.edu/dcaps/dcaps.html进行引物设计,并在F5植株的DNA中进行多态性验证。将F5植株的DNA提取后利用200 μL 1×TE Buffer进行稀释。PCR反应液根据TaKaRa 的PrimerSTAR Max DNA Polymerase试剂盒说明进行配制。PCR反应条件:95 ℃ 10 min;92 ℃ 30 s,52 ℃ 30 s,72 ℃ 1 min,35个循环;70 ℃ 10 min。SSR引物的PCR产物银染后在8%聚丙烯酰胺凝胶上进行电泳拍照记录,dCAPS引物的PCR产物利用对应的酶进行酶切后,在3%琼脂糖凝胶上进行电泳拍照记录。
1.4 花色基因连锁定位
分别与紫花或白花DNA池一致的多态标记分别记为“a”或 “b”,不清楚或偏分离的记为“-”。表型为紫花和白花的分别记为“d”和“b”,利用Join Map 4.1 内置算法Regression algorithm 和 Kosambi函数对进行遗传图谱的构建。运用MapChart 2.2软件进行图谱绘制[19]。
2 结果与分析
2.1 花色性状遗传分析
MS轮回群体的F6于2017年进行夏播,行长6 m,在开花期调查其花色性状,发现1个花色分离的株行。75个单株有58个表现为紫花,17个白花,符合3紫花∶1白花的表型分离比例(P>0.05) (表1);对75个单株衍生的F6∶7家系。2018年种植75个单株衍生的F6∶7家系,结果显示,17个白花的后代株行均表现为白花;58个紫花的后代株行中,20个株行未出现花色性状分离,38个株行出现花色性状分离,符合1紫花∶2花色分离∶1白花的预期分离比例(P>0.05)。结果表明,在本研究群体中大豆花色性状由单基因位点控制,其中紫花对白花为显性性状,研究结果与以往研究结果一致[20]。

表1 大豆花色的遗传分析Tab.1 The genetic analysis of soybean flower color
2.2 重测序变异基本分析
2018年在 MS轮回群体F6∶7的20个和17个纯紫花或白花株行中随机取1株采集叶片,将紫花和白花分别混成样品池,提取其DNA进行高通量测序,测序数据量为10 Gb。对样品池间SNP变异进行比对分析,共检测到329 992个SNP变异位点。由基因功能区分布可知(图1-A),其中252 651个(占比76.56%)SNP变异位点分布于基因间隔区(Intergenic region)。可变剪切区(Splicing region)中SNP变异位点分布最少,仅为81个(占比0.02%)。此外,3个SNP变异位点分别跨5′UTR和3′UTR区域,4个SNP变异位点位于外显子的可变剪切区。由SNP变异位点在整个基因组和染色体的分布可知(图1-B、C),SNP变异位点在20条染色体上分布相对均匀,占比在4.5%左右。其中,20号染色体富集最多,有32 341个SNP变异位点(占比9.80%),11号染色体最少,仅含有5 186个(占比1.57%)SNP变异位点,此外,不同染色体区段存在多个明显SNP变异位点富集区,表明这些区段可能为大豆花色调控基因的候选区段。但此结果仍无法判断调控大豆花色位点的区间,因此需要进一步筛选。
2.3 花色候选区段筛选
为进一步明确大豆花色调控基因的候选区间,针对原始测序结果进行2步筛选:①选出质量较高的SNP(Quality>100)变异位点;②去除杂合位点。筛选后统计结果如下:共获得3371个高质量且纯合的SNP变异位点。由基因功能区分布可知(图2-A),其中2 424个(占比71.93%)SNP变异位点分布在基因间隔区(Intergenic region),可变剪切区(Splicing region)分布最少,仅为2个(占比0.06%)。由SNP变异位点在整个基因组和染色体分布可知(图2-B、C),SNP变异位点在20条染色体上分布比例为0.98%~20.77%。其中,13号染色体富集最多,有700个SNP变异位点(占比20.77%),表明调控大豆花色的基因最可能位于13号染色体上(图2-C)。进一步分析其富集的区段,结果显示,在13号染色体20~30 Mb的物理区间有显著的SNP富集区(图2-B),表明该SNP变异位点富集区为强候选区间。

图1 筛选前SNP变异位点分布Fig.1 The original distribution of SNP variation before screening
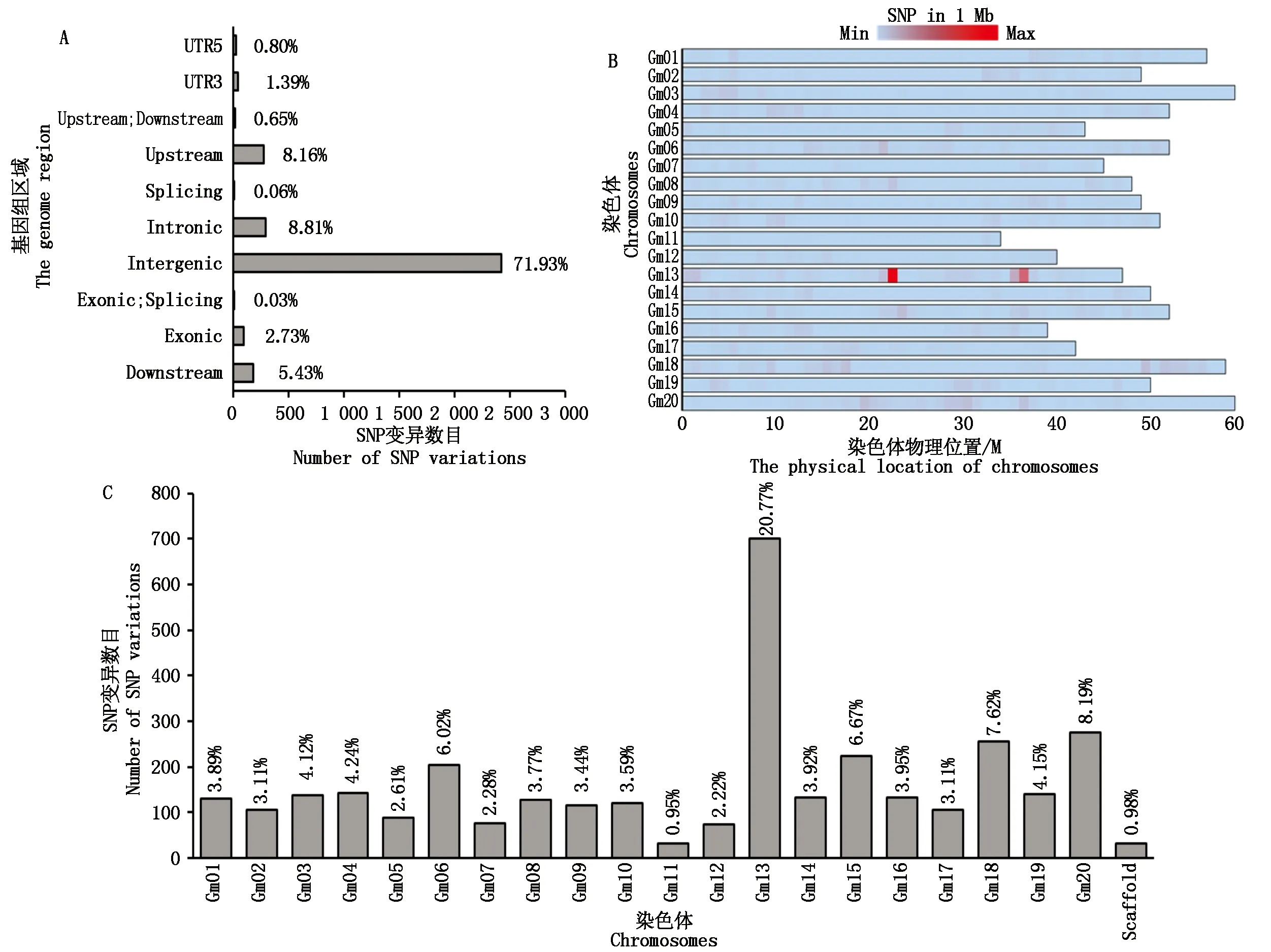
图2 筛选后SNP变异位点分布Fig.2 The distribution of SNP variation after screening
2.4 dCAPs 标记的开发和调控大豆花色基因位点的连锁分析
为进一步证实结果的可信性,在候选区间物理位置21 931 375和21 948 774处分别开发了2对dCAPS标记(表2),同时为了防止设计标记出现异位扩增现象,同时筛选出了2对具有多态性的SSR标记Satt149和Satt516作为参照。进一步利用F6株行的75个单株和4个多态标记进行连锁分析。结果显示(图3), dCAPS-1、dCAPS-2、W1、Satt149和Satt516之间均呈现连锁,遗传距离为Satt149-(8.8 cM)-W1-(0.4 cM)-dCAPS-1-(2.3 cM)-dCAPS-2-(8.7 cM)-Satt516,整个遗传图谱共可覆盖20.2 cM的遗传距离,其中2个dCAPS标记位于W1的同一侧,遗传距离分别为0.4,2.7 cM。结果表明,新开发的dCAPS标记未出现异位扩增,可以代表目标位置的多态性。此外,新开发的标记均与目标性状紧密连锁,且定位W1所在的位置与以往报道一致[21],表明新开发的dCAPS标记可以用于后续的MAS中。

表2 本研究中使用的dCAPS引物Tab.2 The dCAPS primers used in this study
注:引物中下划线的碱基表示引入的突变碱基位点。
Note:The underlined font indicates the position of introduced SNP variation.
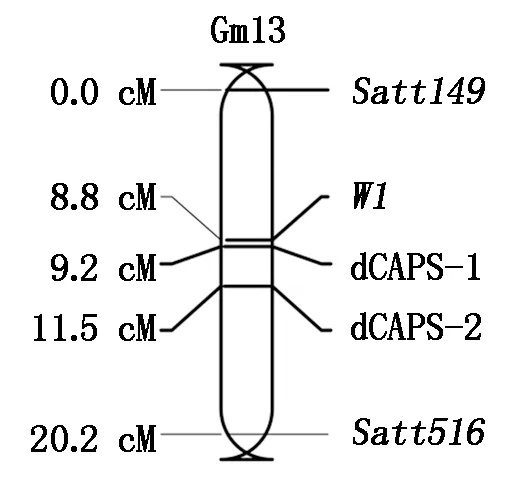
图3 花色位点W1的连锁图谱Fig.3 The genetic map of W1 for flower color
3 讨论
群体分离分析(Bulk segregation analysis, BSA)是一种可用于快速定位质量性状或含有主效位点的数量性状的研究方法,在水稻、小麦、玉米和大豆等作物的遗传定位的研究中被广泛使用[22-24],通常使用F2或RIL群体进行混池的构建。其中F2群体构建比较简单快速,但对于质量性状而言,子代池中显性性状池会混入杂合基因系,对定位效果存在很大干扰;虽然利用RIL群体材料可以很好解决这个问题,但RIL群体构建周期较长,并且对数量性状定位的效果并不十分理想。对数量性状位点的精细定位需要构建更高级的遗传群体,例如剩余杂合系和近等基因系。花色性状在育种过程中是非常容易区分的一个质量性状,尽管没有证据表明对大豆产量具有显著影响,但在品种审定的过程中,花色分离往往会被淘汰。在田间育种的过程中,高世代株行发生花色分离是一种常见的现象,严重影响了育种的效率。本研究从解决田间高世代株行材料发生花色分离的现象入手,将高世代F6∶7中不分离的株行构建成紫花和白花DNA池,采用重测序的办法进行定位研究。由于使用了高世代材料,材料间遗传背景相似度高,材料间属于近等基因系,极大排除了遗传背景的干扰,易于鉴定。结果也显示,筛选出的SNP标记形成了明显的富集区,在富集区开发的dCAPS标记可以很好地追踪花色性状,表明这2个标记可以应用到分子标记辅助育种当中。
大豆是古四倍体作物,约在5 900万a和1 300万a之前经过2次染色体加倍事件[25],随后经历染色体丢失、重排、分化等事件逐渐形成现代的2倍体大豆。因此,大豆基因间序列具有高度的同源性,因此一个基因可能存在多个拷贝的现象。而不同拷贝间的变异又是相对独立的,因此不同拷贝间也存在变异。目前主流的重测序技术是二代重测序,主要以在50~250 bp的DNA片段加接头的方式进行高通量的测序,获得原始数据与参考基因组进行比对分析来确定遗传变异[26]。但受限于参考基因组拼接质量和基因组中多拷贝变异之间的影响,往往会鉴定出大量假阳性的SNP变异位点,这种假阳性率在大豆、小麦等古四倍体或多倍体作物中尤为高。而一般测序比对默认的筛选参数较低[27],因此加大了假阳性出现的概率。在研究中发现,根据测序通用标准在紫花和白花2个混池中仅鉴定到约33万个SNP变异位点,说明从F6∶7株行的植株间遗传背景已经非常相近。但这33万个SNP变异位点未呈现明显的富集区,不能明确调控花色位点的准确遗传位置,推测存在较多的假阳性位点。因此,通过提高测序质量(Quality 参数),进一步提高位点真实性。此外,构建紫花和白花使用的单株均为花色纯合单株,因此与目标位点连锁的区域也趋于纯合,因此进一步将呈现杂合的位点排除。最终得到3 371个候选位点,并且在13号染色体处形成了一个明显的富集区,说明未筛选前数据确实存在大量假阳性的SNP变异。
本研究利用田间高世代育种材料和高通量测序技术,对大豆花色调控基因位点W1进行了快速定位。研究结果显示,利用高世代分离材料可用于有效、快速排除遗传背景的干扰,提高了遗传定位效率,在W1位点候选区间内开发的分子标记可以有效地跟踪W1的遗传走向,研究结果为今后的MAS育种提供了理论参考基础。
