CRISPR / Cas9系统介导GFP在兔H11位点的定点整合研究
2019-10-24姚洋尤双刘芝瑾张翔宇侯晓旭郭涛倪伟胡圣伟
姚洋,尤双,刘芝瑾,张翔宇,侯晓旭,郭涛,倪伟,胡圣伟
(石河子大学生命科学学院,新疆 石河子 832003)
家兔具有繁殖周期短、易进行超数排卵、与人类生理结构相似等特点。目前,转基因家兔已经广泛用于人类疾病模型的研究[1],包括脂类代谢和动脉硬化,以及眼科和整形外科等几个领域[2]。转基因家兔也被用于生产稀有药用蛋白[3],相关药物已经在美国和欧盟批准上市。但是,目前转基因家兔的外源基因都为随机整合,导致外源基因表达存在不稳定和表达量低等问题,这在一定程度上限制了转基因家兔在生物医药领域的应用。因此,研究家兔外源基因定点整合技术及友好位点,对定点整合转基因兔的制备具有重要意义。
使用工程化核酸酶的靶向基因组编辑已经成为多数生物学研究使用的主流方法[4]。目前已用于基因功能的研究,提供用于生物和医学研究的转基因动物模型并改善畜牧业中动物的特定性状。定期间隔短回文重复序列(CRISPRs)和Cas蛋白是跨细菌和古细菌的广泛系统,其导致对外来核酸的干扰[5]。该系统由20nt的指导序列(sgRNA)和Cas9核酸酶组成,利用非同源末端连接(NHEJ)来诱导靶标突变。将sgRNA和Cas9 mRNA同时注射受精卵[6],已成为基因靶向动物模型的重要工具。
传统上,通过将基因序列或转基因以随机方式整合到基因组中来产生转基因动物,其中转基因可以插入基因组中的任何位置[7]。转基因的随机整合经常导致不稳定的表型,基因沉默和不可预测的基因表达,并且在一些情况下,该过程是诱变的。对于位点特异性基因插入,过去使用同源重组(HR)和体细胞核移植(SCNT)以产生转基因动物[8]。
由于HR效率低,费力且耗时,这种方法效率低,所以急需一种高效的方法解决这种问题,基因定点整合技术利用同源重组和基因编辑为解决这种问题提供了优良的方法。2014年,赖良学的研究团队成功获得了世界上首个Rosa26定点基因敲入猪的模型。2015年,Zambrowicz等人结合同源重组和基因编辑技术,得到了基因敲入兔[9]。
友好位点是表达比较活跃的靶位点,其具有高整合效率,稳定表达和高插入基因表达的优势。Hipp11(H11)位点由HIPPENMEYER[10]最早发现,随后在基因敲入小鼠[11]和人类干细胞[12]中进行了验证。在小鼠基因组中,H11位点处于Eif4enif1和Drg1基因中间。然而在兔子中H11位点开发却没有报道。本研究通过比对小鼠H11位点,在家兔基因组中开发出兔的相应H11位点。并且利用CRISPR/Cas9介导基因定点整合方法将GFP基因整合到家兔H11位点上。
通过对CRISPR / Cas9系统在兔中的定点整合进行鉴定,可以很好的解决随机整合带来的外源基因表达的不均一等问题,使插入的外源基因能够高效率的表达;同时,使外源基因定点整合的效率极大的提高,生产转基因兔的成本大大降低,这为未来制备转基因兔搭建了一个安全、高效的平台。
1 材料与方法
1.1 主要试剂及仪器
Trizol试剂、胎牛血清、DMEM培养基、无血清培养基、青霉素-链霉素溶液均购于美国赛默飞世尔科技公司;DL10000DNA marker、DL2000DNA marker、DL1000DNA marker(宝生物工程公司);限制性内切酶BglⅡ、AseI、NheI、AflⅡ和BbsⅠ,Universal Genomic DNA Extraction Kit Ver.5.0购于北京Takara公司;胰蛋白酶、氯化钠、酵母提取物、琼脂粉,琼脂糖均购自索莱宝公司。
Lonza4D-Nucleofector电转仪(Lonza,公司,德国)、冰箱(海尔,中国),低温离心机、倒置荧光显微镜系统(Nikon,德国)、移液器(Eppendorf 公司,德国),超净工作台(苏州安泰空气技术有限公司,中国),二氧化碳细胞培养箱,琼脂糖凝胶核酸电泳仪(北京六一生物科技有限公司,中国);超低温冰箱(三洋公司,日本),生化培养箱(上海跃进医疗器械有限公司,中国),恒温水浴锅(上海精密仪器仪表有限公司,中国),生物安全柜(BAKER公司,美国),立式压力蒸汽高压灭菌器(上海博讯公司,中国),凝胶成像系统(伯乐公司,美国)。
1.2 序列比对
以EIF4ENIF1或Drg1基因为标识,在NCBI 网站查找小鼠、人和猪的 H11 位点及其周边序列,并预测兔的 H11 位点的位置和周边序列,与猪的序列利用 DNAMAN进行比对分析。
1.3 CRISPR/Cas9敲除载体的构建
利用CRISPR/Cas9在线设计工具(http://tools.genomeenging.org)根据预测的H11位点和周边序列,以DNA正义链(反义链)的序列GGN18NGG(GGN17NGG)为靶位点设计两条sgRNA,引物见表1。取100 μmol/L的sgRNA上游和下游引物各10ul,放置于95 ℃水浴5 min后,关闭水浴锅,在水浴中冷却至室温,退火,获得带有粘性末端的双链CRISPR/Cas9。
同时利用BbsⅠ线性化 PX330载体。线性化的 PX330载体经1%琼脂糖电泳检测,胶回收后,线性化的PX330载体和退火的sgRNA用T4连接酶连接过夜,经转化后,挑取单克隆送睿博兴科公司测序。

表1 sgRNA序列设计Tab.1 SgRNA sequence design
1.4 同源打靶载体的构建
pIRES2-ZsGreen1-Puro(本文缩写为pIRZP)载体已由本实验室构建保存。试验依据兔H11位点两侧的左右同源臂序列(图1)分别设计合成左右同源臂的引物,在H11位点左右同源臂引物的5′端分别添加AseI(ATTAAT)和AflⅡ(TTAAG)酶切位点。利用H11-3′Arm-F和H11-3′Arm-R引物通过PCR扩增、酶切、连接,将右同源臂克隆至pIRZP载体中,经测序验证,得到pIRZP-H11-3′Arm载体。PCR反应体系如下:上下游引物(10 μmol/L)各1 μL,2 × Es Taq Master Mix25 μL,家兔基因组2 μL模板,ddH2O 21 μL。PCR 反应程序:94 ℃,5 min;94 ℃,30 s,55 ℃,30 s,72 ℃,50 s,共35 个循环;最后 72 ℃,5 min;4 ℃保存。
用上述PCR的方法利用H11-5′Arm-R和H11-5′Arm-F引物将H11位点左同源臂克隆至pIRZP-H11-3′Arm载体上,送睿博兴科公司公司测序,引物见表2。
1.5 嘌呤霉素筛选兔胚胎成纤维细胞浓度确定
我们将接种于12孔板的兔胚胎成纤维细胞培养24 h后,换用不同浓度的嘌呤霉素细胞培养液继续培养[13]。每孔做3个生物学重复,隔 1 d 观察细胞的生长情况。
至3 d 后,取细胞全部死亡的板孔所对应的最小浓度作为最佳筛选浓度。
1.6 电转染
待6孔板4个孔中的细胞长至70%~80%后,收集细胞,计数。
按照 Lonza Nucleofector的操作说明,各取2×105~1×106个细胞,分别添加如表3的体系之后和 100 μL电转液混合。选择EH-100程序进行电转。电转结束,快速取出电转杯并静置10 min。把电转的细胞转入至温育的含有15% FBS培养基的6孔板中,并于37 ℃,5.0%CO2培养箱中培养,5~6 h后换液。转染24 h后,换最佳筛选浓度的嘌呤霉素培养液对细胞进行筛选。间隔2~3 d,换一次嘌呤霉素培养液,直至长出大而漂亮的克隆岛。

表3 电转体系Tab.3 Electric transfer system
1.7 打靶细胞的检测
我们将敲除载体与同源打靶载体共转染细胞24 h后,利用Universal Genomic DNA Extraction Kit Ver. 5. 0(Takara)提取2种细胞的基因组DNA,之后通过PCR技术检测打靶细胞的GFP基因和Puro基因,PCR反应体系如下:上、下游引物(10 μmol/L)各0.5 μL,2 × Es Taq Master Mix10 μL,细胞基因组1 μL模板,ddH2O 8 μL。PCR 反应程序:94 ℃,5 min;94 ℃,30 s,57 ℃,30 s,72 ℃,40 s,共 35 个循环;最后 72 ℃,5 min;4 ℃保存。与此同时用5’arm-cmv-F、5′arm-cmv-R和GFP-3′arm-F1、GFP-3’arm-R1引物对打靶细胞DNA进行PCR验证。
2 结果与分析
2.1 猪与小鼠与人的 H11 位点周围环境序列比对
将猪与小鼠和人的 H11 位点序列进行比对,从比对结果中看出人、小鼠与猪中Eif4enif1基因和Drg1 基因距离相近且相邻(图2)。并且H11 位点都处于Eif4enif1和Drg1之中,所以我们预测家兔的H11位点也处于Eif4enif1和Drg1之间,随后从Genebank 调出Eif4enif1 和Drg1之间的序列,运用DNAMAN软件对其进行序列的比对,发现人,小鼠和家兔之间的同源性约为 49.67%(图3),红色方框标记H11位点的区域。
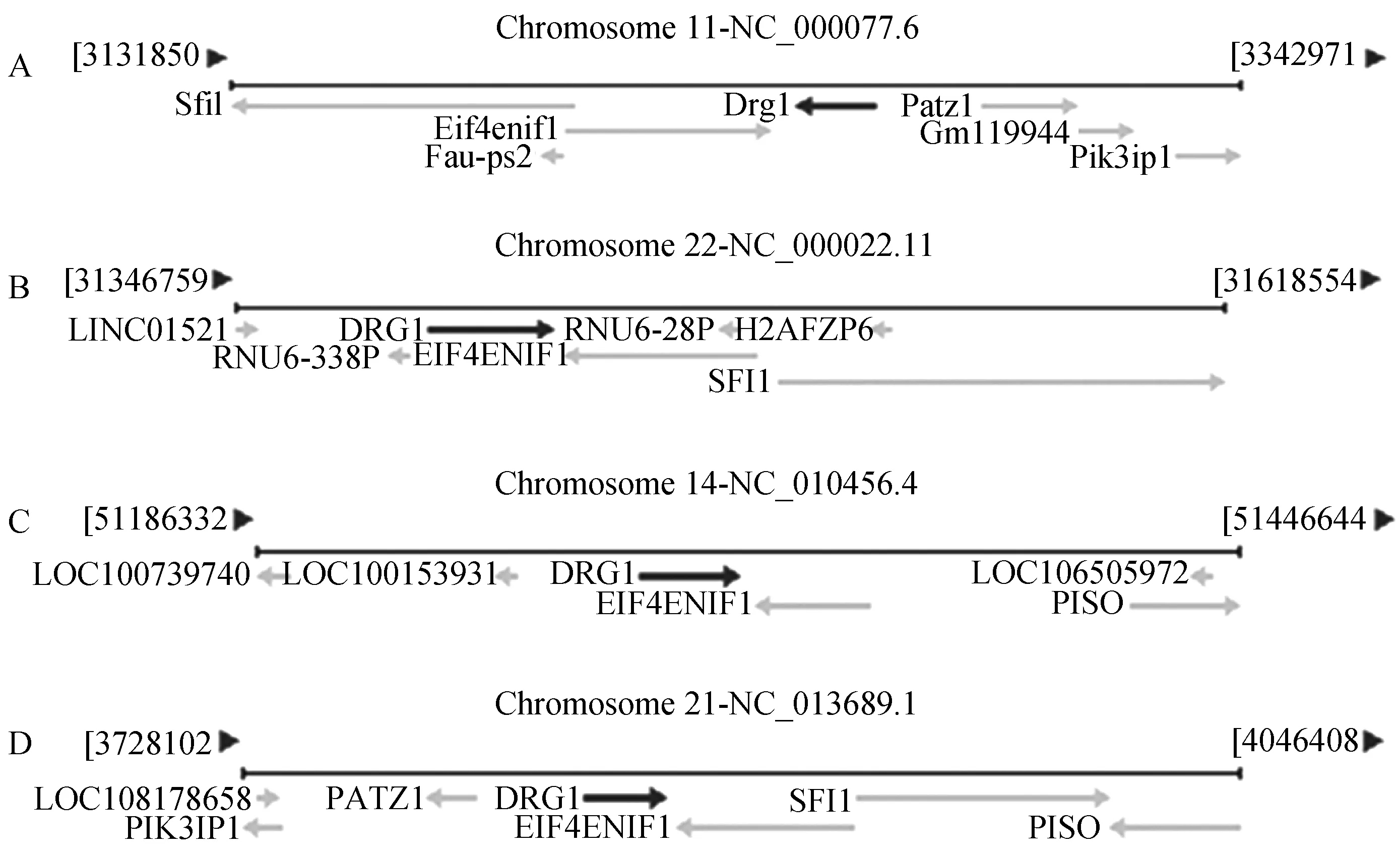
A—小鼠;B—人;C—猪;D—兔图2 H11位点在小鼠、人、猪和兔中的位置Fig.2 Mapping of H11 locus in mice,human,pig and rabbit

图3 小鼠、人、和兔子的H11序列比对Fig.3 Alignment of H11 locus between mice,human and rabbit
2.2 CRISPR/Cas9敲除载体的构建
PX330载体经BbsⅠ酶切后,结果显示与对照相比酶切完全,条带单一(图4)。构建的CRISPR/Cas9敲除载体经过测序验证,结果如图所示sgRNA成功连入PX330载体中(图5),H11-g RNA1和H11-g RNA2敲除载体构建成功。为了检测构建的CRISPR/Cas9敲除载体是否有活性,将上述H11-g RNA1和H11-g RNA2载体转染兔胚胎成纤维细胞。3 d后收集细胞并提取细胞基因组,经T7E1酶切,结果如图6显示H11-g RNA1和H11-g RNA2敲除载体具有生物学活性且效率有2%~10%。

对照:PX330质粒对照;1-8:BbsⅠ酶切产物;M:10000的marker图4 PX330酶切Fig.4 Digestion of PX330
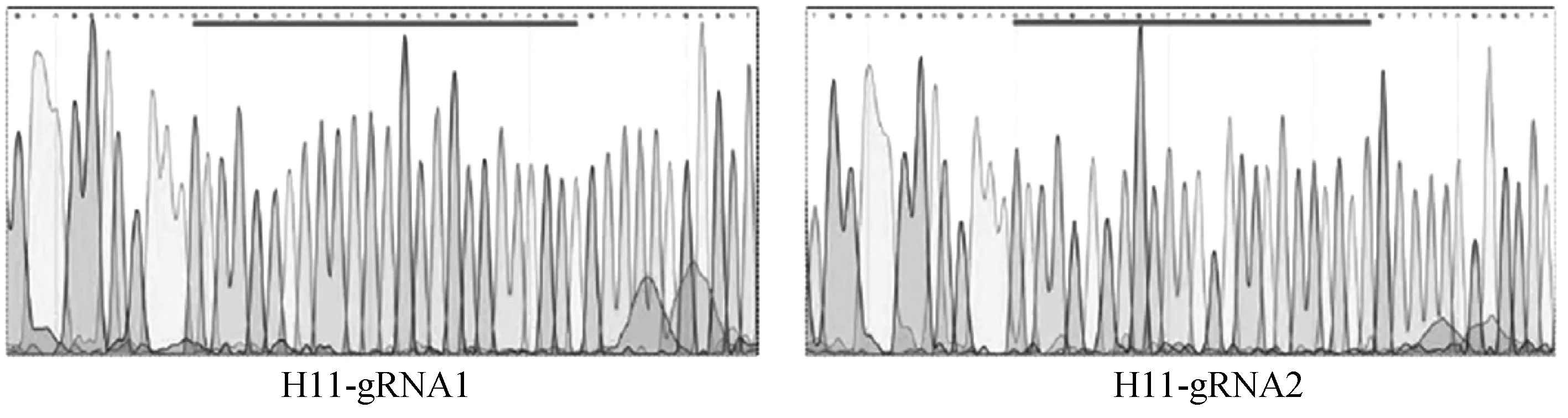
图5 敲除载体的测序结果Fig.5 Knocking out the sequencing results of the vector
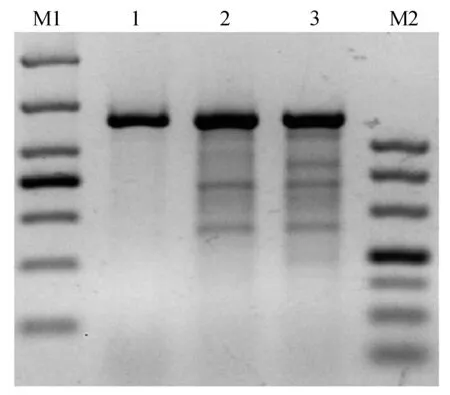
M1:DL1000;1-3:对照,gRNA1,gRNA2;M2:DL500图6 敲除载体T7E1酶切验证Fig.6 knockout vector T7E1 digestion validation
2.3 同源打靶载体的构建
将构建的pIRZP-H11-3’Arm载体用BglⅡ限制酶对其右同源臂的方向进行酶切验证,经电泳分析发现在3号约1658 bp处可以看见清晰的条带,说明3号右同源臂的方向正确(图7)。若右同源臂的方向不正确,则会得到2219 bp大小的片段。进一步对3号质粒测序验证,结果显示H11的右同源臂成功连接到pIRZP载体上。之后对构建的pIRZP-H11载体用PCR验证其左同源臂的方向,若方向正确如图8 A,以H11-5’-F,Puro-R进行PCR就可以扩增出1987 bp片段,而以H11-5’-F,Green-R PCR后可以扩增出3323 bp的片段。若方向错误则不会扩增出条带。测序结果显示H11的左同源臂连接到pIRZP-H11-3’Arm载体上(图8B)。成功构建pIRZP-H11同源打靶载体如图9 A,9B所示。进一步酶切验证pIRZP-H11载体,因为pIRZP-H11载体上有3个AflⅡ酶切位点,2个BglⅡ酶切位点,所以用AflⅡ酶切则会出现3532 bp,3132 bp,717 bp三条清晰的条带,其中717 bp为右同源臂的的大小如图9C第3道。而用NheⅠ和BglⅡ酶切 pIRZP-H11载体会出现5117 bp,1658 bp,606 bp3个条带,其中606 bp为Puro基因的大小如图9C第4道。用AseI酶切pIRZP-H11载体出现6621 bp和793 bp2个条带,其中793 bp为左同源臂的大小。以上结果进一步证明我们构建的pIRZP-H11载体是正确的。
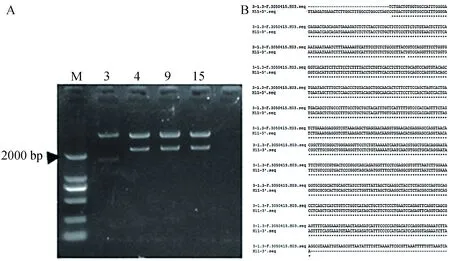
A—酶切验证方向(M:DL2000);B—3号质粒测序结果图7 pIRZP-H11 -3’Arm载体验证Fig.7 pIRZP-H11-3'Arm vector validation

A—PCR验证方向(M1:DL2000;M2:DL10000;1、2:Puro引物;3、4:Green引物;1、3为同一个样品;2、4为同一个样品);B—8号质粒测序结果图8 pIRZP-H11载体验证Fig.8 pIRZP-H11 vector validation
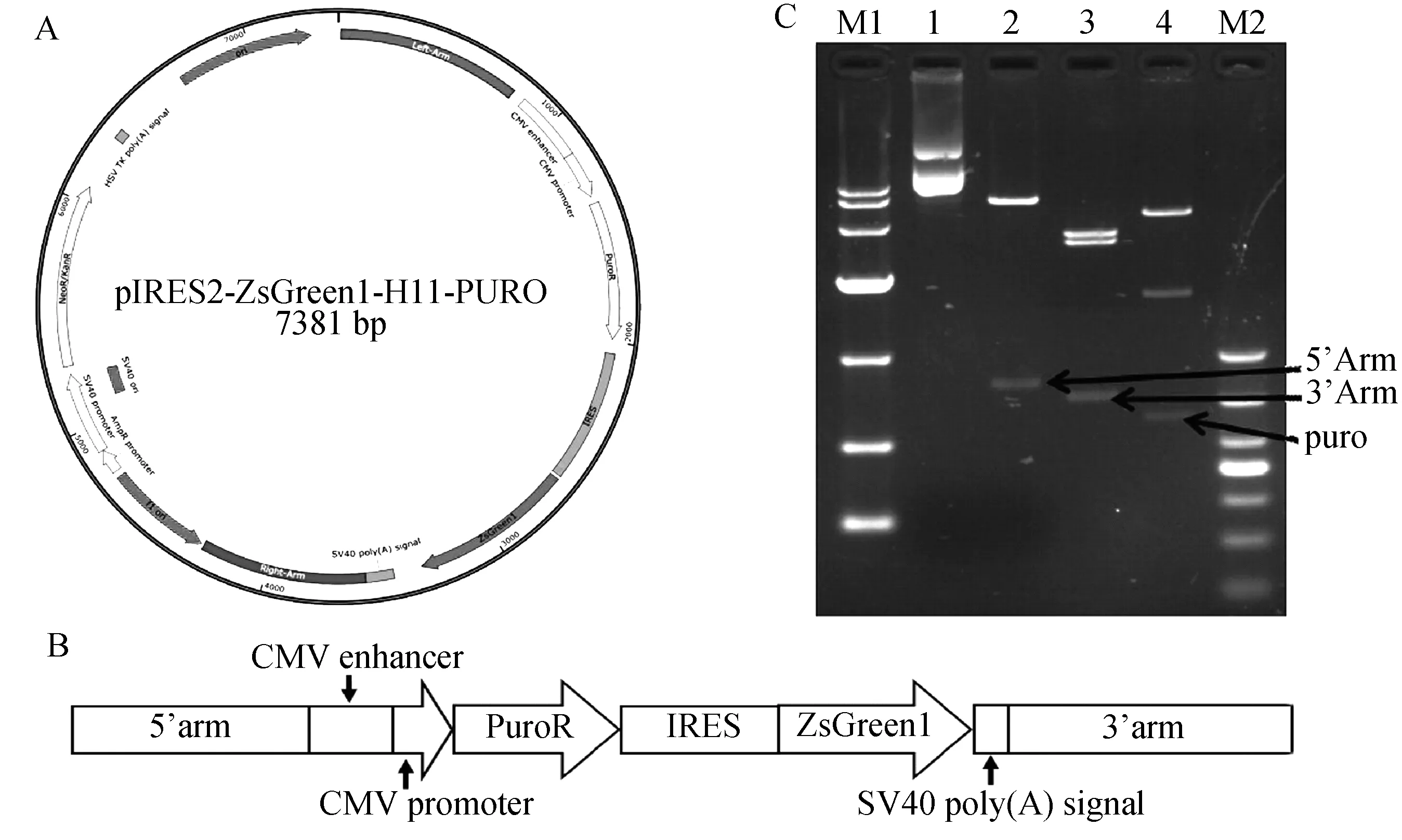
C(M1:DL10000;1-4:对照,Ase I酶切,BglⅡ和NheⅠ酶切,AflⅡ酶切;M2:DL1000)图9 pIRZP-H11载体图和pIRZP-H11载体酶切Fig.9 pIRZP-H11vector and Digestion of pIRZP-H11 vector
2.4 嘌呤霉素最佳浓度筛选结果
经过不同浓度嘌呤霉素的筛选,最终我们确定了在兔胚胎成纤维细胞中,嘌呤霉素的最佳筛选浓度为2 μg/mL。在该浓度下,兔胚胎成纤维细胞经过3天的筛选就已全部死亡。
2.5 GFP表达及药物筛选单克隆群
将敲除载体和打靶载体共转染兔胚胎成纤维细胞24 h后,在40X倍镜下观察到共转染后兔胚胎成纤维细胞形态良好,而荧光表达量和细胞数量均比较少。我们还发现H11-gRNA1+ pIRZP-H11共转染兔胚胎成纤维其荧光表达量和细胞状态高于H11-gRNA2+pIRZP-H11,在此基础上进行后续嘌呤霉素抗性筛选研究。当兔胚胎成纤维细胞共转染H11-gRNA+ pIRZP-H11质粒经过嘌呤霉素筛选到25 d时,在显微镜下可以观察到如(图10)所示的大的具有绿色荧光以及Puro抗性的单克隆群。分别在共转染H11-gRNA1+ pIRZP -H11和H11-gRNA2+ pIRZP-H11质粒的兔胚胎成纤维细胞中找出9个和8个相对大的克隆群(图11)。
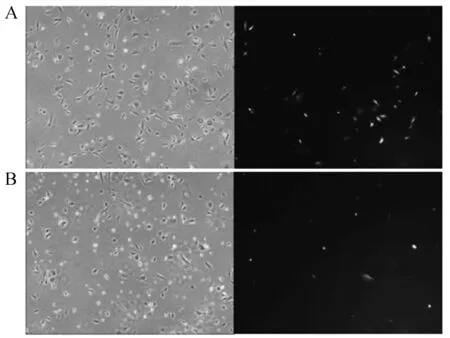
A—H11-gRNA1+ pIRZP-H11;B—H11-gRNA2+ pIRZP-H11图10 共转染兔胚胎成纤维细胞24 h荧光图(40×)Fig.10 Co-transfected rabbit embryonic fibroblasts for 24 hours(40×)

A—H11-gRNA1+ pIRZP-H11;B—H11-gRNA2+ pIRZP-H11图11 兔胚胎成纤维细胞药杀25 d 单克隆荧光图(40×)Fig.11 Rabbit embryonic fibroblasts were killed by 25 days(40×)
2.6 打靶细胞的检测
提取共转染后兔胚胎成纤维细胞的基因组,通过PCR检测后如图12A,可以看到Puro基因和GFP基因清晰单一的条带且大小与我们预期的相同,这说明共转染的兔胚胎成纤维细胞基因组中已存在Puro和GFP基因,进一步用5’arm-cmv-F,5’arm-cmv-R和GFP-3’arm-F1,GFP-3’arm-R1两对引物对打靶细胞基因组定点整合做PCR鉴定,如图出现预期大小的单一条带(图12B)以上结果证明这两个基因已经成功整合至兔胚胎成纤维细胞基因组中。
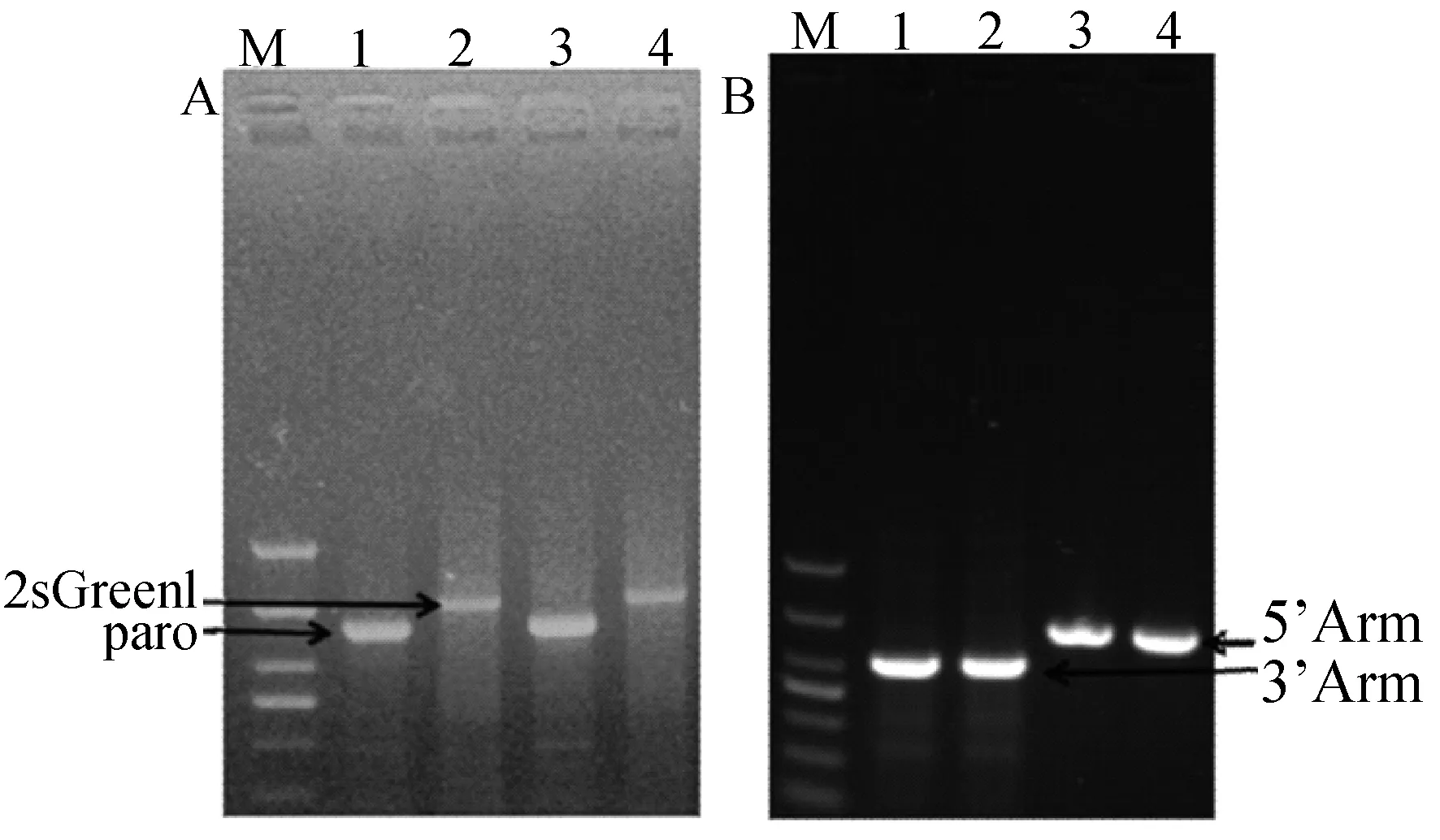
A—M:DL1000;1、2:共转染H11-gRNA1+ pIRZP-H11质粒细胞的Puro和ZsGreen1;3、4:共转染H11-gRNA2+pIRZP-H11质粒细胞的Puro和ZsGreen1;B—M:DL2000;1、3:共转染H11-gRNA1+pIRZP-H11质粒细胞左、右同源臂的junction;2、4:共转染H11-gRNA2+pIRZP-H11质粒细胞左、右同源臂的junction图12 打靶细胞的PCR检测和junction PCR检测Fig.12 PCR detection of target cells and detection of the transfected cells
3 讨论
1985年,Hammer等人研究出世界上首例转基因兔[14]。此后,在全世界内,具有不同研究目的的转基因兔模型相继被生产出来,例如淋巴细胞白血病肿瘤和乳头状瘤[15],获得性免疫缺陷综合征(AIDS)[16]和药用蛋白反应器的研究。然而,大多数转基因兔模型是由外源基因通过原核显微注射被随机插入到兔基因组中产生的。在这过程中存在一些问题,因为外源基因插入到兔基因组内的随机位点上,所以转基因会受到位置效应[15-16],例如:转基因可能会通过插入进而诱变和破坏兔本身基因的功能,因此,科研人员为了筛选足够多的转基因的表达系统并且获得可重复的结果,他们一般运用多条线进行筛选[17]。
为了建立有效的基因敲入系统,本试验首先研究了3种基因组编辑方法:锌指核酸酶(ZFNs)[18],转录激活因子类似效应因子核酸酶(TALEN)[19]和RNA 指导的 CRISPR/Cas核酸酶系统。研究显示Cas9 与 ZFN 和TALEN 相比,有一些潜在优势,包括制作简单,低成本,高效性和促进多重基因组编辑的能力。JIN[20]等人的数据显示,与CRISPR / Cas9n或TALEN相比,CRISPR / Cas9在猪H11基因座上靶向和切割DNA的效率更高,具有高达50%~60%的靶向效率。本试验建立的基因敲入系统将GFP和Puro基因高效的整合进入兔的H11基因座上,再一次证明了CRISPR / Cas9基因编辑技术的优越性。
本试验首次证明了CRISPR / Cas9可以有效地用于产生兔模型。在本试验中,通过将CRISPR / Cas9、同源定向重组(HDR)和兔子H11位点结合构建成一套系统,在兔中实现精确的,位点特异性的基因整合及表达,这是一种新的策略。使用CRISPR / Cas9,可以实现高达较高的基因整合效率,报告基因GFP和Puro已存在于兔的基因组中。相比于随即整合方法,本试验的方法有许多优势,如:去除了位置效应,有比较稳定的单拷贝插入以及高整合效率[21]。用传统HR 的目标转基因通过药物筛选的效率低于 6%,由于其许多元件和大片段,传统HR 基因靶向载体的构建,在技术上有挑战性且耗时[38-39]。
H11 位点,位于Eif4enif1基因 与Drg1 基因中间,而这两个基因在不同种属的位置有所差异,在小鼠、人、猪中分别位于第11 号、22 号、14 号染色体,而在兔中位于第21号染色体,该位点不含有任何启动子[22],在具有转录活性的H11位点插入外源基因,可以实现通过启动子例如CMV驱动的基因表达[20]。在以后的克隆过程中,将该敲入系统用于兔胚胎成纤维细胞中,可以在短时间内获得具体的任何所感兴趣的基因插入的转基因兔。此外,本试验的敲入系统有效的敲入 1.2 Kb 片段,有研究称可插入 4.2 Kb,甚至高达9.4 Kb的大片段[20],由此可见使用CRISPR / Cas9系统,片段的大小对基因插入的效率似乎没有明显的影响。CRISPR / Cas9介导的同源定向重组基因编辑可以成为精确和高效的获得转基因兔的有效手段,相信在未来,该敲入系统能够在大型物种转基因动物的生产中发挥一定的作用。
4 结论
试验运用CRISPR/Cas9 基因编辑系统获得敲除载体。针对H11位点,试验利用基因的HDR构建了同源重组载体,在兔胚胎成纤维细胞系中建立了一个实验平台,该平台运用HDR机制进行了基因的靶向敲入。通过该平台获得了较高重组效率的兔胚胎成纤维细胞,这为以后试验打下坚实的基础。结合荧光和药物筛选以及分子生物学手段,成功的在兔基因组中检测我们期望的外源基因,说明外源基因已整合到基因组中,且HDR机制的定点整合是可行的,为后续试验奠定基础。
