掺Ag浓度对ZnO(100)面吸附S的电学特性影响
2019-10-11朱婷
朱婷
(西南石油大学理学院,成都610500)
1 引言
ZnO 作为一种宽禁带的半导体材料,室温下其带隙为3.37Ev。与大多数的宽禁带半导体(如GaN、ZnSe、ZnS 等)相比,ZnO 的激子束缚能为60meV,高于室温热离化能,有利于激子在室温下实现高效的激子发射[1]。此外,ZnO 的硬度大、介电常数低、光电耦合率高、热学及化学稳定性强,因此,在压敏器件、气敏传感器件、紫外光电探测器,以及太阳能电池等方面具有广阔的应用前景。
本文以Ag 掺杂ZnO 为研究对象,采用密度泛函理论的第一性原理系统地模拟计算掺Ag 浓度对ZnO(100)面吸附S的电学特性影响,揭示了吸附能改变的内在机理,为其在脱硫吸附技术的研究提供了一定的参考。
2 理论模型与计算方法
2.1 构建模型
本文采用ZnO 的立方闪锌矿结构作为基本的晶格模型。首先,对掺杂前的ZnO(100)面吸附S 构建模型,将一个ZnO原胞在x,y 方向上分别扩展一个单位得到2×2×1 的ZnO 超晶胞(包含8 个Zn 原子与8 个O 原子)。在ZnO(100)面分别选取四种不同的S 原子吸附位置:Zn-Zn 键位的正上方、四方Zn原子结构的正上方,以及周期性排列的两个相应位置。随后,对掺Ag 后的ZnO(100)面吸附S 构建模型,这里采用的掺杂方式为替位掺杂。在以上的四种吸附位置中选取最稳定的位置作为研究对象,将ZnO 晶胞中的Zn 原子替换为Ag 原子。
2.2 计算方法
采用CASTEP 软件包分别对掺Ag 前后的ZnO 超晶胞进行计算。计算前,先对建立的晶体模型进行几何优化,使其达到最稳定的结构。在Kohn-Sham 能量泛函形式中,选择广义梯度近似(GGA)的PBE 处理电子间的交换关联能,采用超软贋势平面波选择基函数,采用1×1×1 的Monkorst-park 特殊K点对全Brillouin 求和,整个计算都在倒易空间中完成。具体的参数设定如下:平面波截断能量设定为Ecutoff=300eV,自洽收敛能的精度为2×105eV/atom,最大位移为0.002A°,晶体内应力收敛标准为0.05GPa,原子的相互作用力收敛标准为0.05eV/A°。
3 计算结果和分析
3.1 掺Ag 前ZnO(100)面吸附S 的电学特性
为比较上述四种不同吸附位置下的吸附稳定性,分别计算各自的吸附能为:ΔEmd=(ES+EA)-ET
其中,ES、EA和ET分别表示吸附前ZnO(100)面的能量,S原子本身的能量,以及吸附后体系的总能量。经计算得知EA=-273.9321eV,ES=-3.4350×104eV。上述四个吸附位置的体系总能量和吸附能如表1所示。由于在吸附过程中释放的吸附能越大则体系越稳定,因此,Zn-Zn 正上方的吸附位置具有最稳定的吸附性。
为反映吸附位置对ZnO(100)面导电性的影响,分别计算各自的能带结构。可以看出,导带底与价带顶对应的是同一K值,因此ZnO 是直接带隙导体。对于四种不同的吸附位置,ZnO(100)面的能带带隙体现了较为显著的差异,可得出,S 原子吸附的位置越稳定,ZnO(100)面的能带带隙越大,于是其(100)面的导电性越弱,电阻率越大。
在S 原子的四种吸附位置下,计算ZnO(100)面的电子态密度,如图1所示。可以发现,相比于其他吸附位置,位置A(具有最稳定的吸附性)的s 态与p 态密度分布均体现了较为显著的变化。ZnO(100)面的电子态主要分为三个区域:-22.5~-17.5eV 的价带,-13~-11eV 的下价带,以及-7.5~6eV的上价带。s 态密度与p 态密度分别在价带与上价带区的峰值最高。很大程度上,三个区域分别来源于O 的s 态电子,Zn 的d态电子,以及O 的p 态电子的贡献。可以得出,S 原子吸附在ZnO(100)面的不同位置时,对称位间的态密度并无太大的差异。
3.2 掺Ag 后ZnO(100)面吸附S 的电学特性
根据以上S 原子吸附位置对ZnO(100)面导电性的影响分析,选择吸附最稳定的位置为对应的ZnO 为研究对象,对ZnO 进行替位掺Ag。结合ZnO(100)面能带带隙随掺杂浓度变化的模拟曲线(如图2所示,掺Ag 浓度分别标记为E、F、G、H),随着掺杂浓度在以上范围内的增大,能带带隙呈先减小再增大的变化趋势,并且在的浓度区间内出现极小值,此时ZnO(100)面的导电性最强。

表1 各吸附位置的吸附能参数
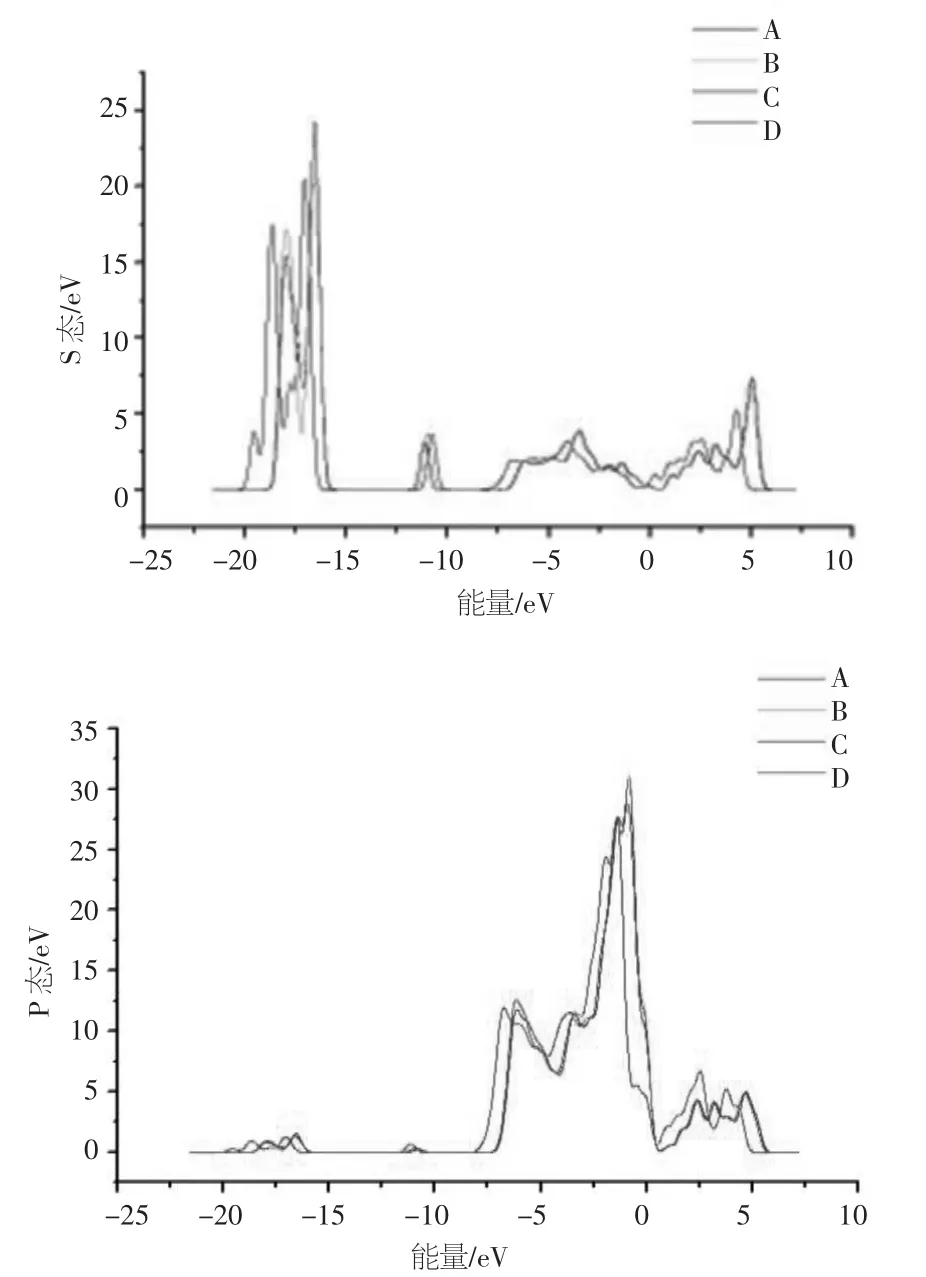
图1 掺Ag 前ZnO(100)面吸附S 的分波态密度图

图2 ZnO(100)面能带带隙随掺Ag 浓度的变化关系
图3显示了掺Ag 后ZnO(100)面的分波态(s 态与p 态)密度。由此看出,ZnO(100)面的电子态主要分为三个区域:-22.5~-17.5eV,-13~-11eV 以及-7.5~6eV。其中,在-13~-11eV与-7.5~6eV 两个区域里,s 态密度明显减小,而p 态密度则显著增大。结合掺杂前的分波态密度图可以发现,随着掺Ag 浓度的增大,导致价带区的p 态电子密度增大,而s 态电子密度减小,从而使ZnO(100)面的导电性增强。

图3 掺Ag 后ZnO(100)面吸附S 的分波态密度图
4 结论
本文采用基于密度泛函理论的第一性原理方法,在不同的掺Ag 浓度下对ZnO(100)面吸附S 的电学特性进行了计算分析。
结果表明:①ZnO(100)面吸附S 释放的吸附能越大,则吸附性越稳定,于是确定Zn-Zn 键的正上方作为S 原子的最佳吸附位置,此时ZnO(100)面的能带带隙最大,因此,其导电性最弱。②以上述的吸附位置为研究对象,对ZnO 进行替位掺Ag,随着掺Ag 浓度的增大,ZnO(100)面的能带带隙并无显著的变化,而价带区的p 态电子密度明显增大,s 态电子密度减小,因此,ZnO(100)面的导电性增强。
