药物代谢动力学经典模型的局限与改进
2019-09-10曹永孝
曹永孝
(西安交通大学医学部药理学系,陕西西安 710061)
药物代谢动力学(pharmacokinetics)主要研究药物的体内过程,定量阐述药物的体内动态规律,为制订给药方案提供依据,是药理学的重要组成部分,对新药研究开发、药物合理使用和药品质量控制有重要理论和实际价值。
药物代谢动力学的历史可追溯至1841年,苏格兰学者ALEXANDER[1]进行了第一个人体药物代谢实验。1924年,瑞典学者WIDMARK及TANDBERG[2]提出了开放性一室模型的概念,为房室模型奠定了基础。1937年,TEOREU[3-5]提出两室模型,是现代药物代谢动力学理论奠基人。1953年,DOST[6]首次在教科书中提出了药物代谢动力学的概念,数百位科学家随后进一步研究对其发扬光大。1960年,BRODIE[7]阐明了药物通过细胞膜的机制及药物体内分布规律,建立了药物代谢动力学模型及其参数的计算方法。1978年,YAMAOKA[8]用统计矩原理,将非房室分析法应用到药动学研究,目前已成为药动学研究的主流方法,也是各国药品审评当局推荐使用的药动学研究方法。我们对2017年Pubmed发表的有关药物药动学研究的文献进行统计分析发现,统计矩模型的应用达90%。房室模型和统计矩模型均是药代动力学的里程碑事件,是生理药物动学模型、药动-药效组合模型、群体药动学等模型的基础,为药物代谢动力学的发展做出了突出贡献。然而,任何研究都有局限性,任何模型、方法都不可能完全模拟人体的状况,甚至会有很大的差距。认识这些差距,是发现新途径和新方法的前题。
1 模型的基本思想
药物的体内过程复杂,同一药物于同一时刻在不同器官的量不同,同一器官的不同时刻药量也不相同,因而完全弄清药物在体内的分布非常困难。为此,科学家相继提出了不同的模型和理论,房室模型和统计矩模型是药物代谢动力学的经典模型和基本理论。
1.1 房室模型房室模型将人体的组织器官按药物的分布速率假设成不同的房室,分布速率相近者视为同一室,进入机体的药物按分布速率分布于房室中[9]。一室模型认为,药物进入机体后,迅速分布到各器官,达到平衡,其对数药物浓度-时间曲线为直线。对于二室模型,药物先随血流分布到血流速度大的器官,而后快速向周边室分布,同时缓慢消除。对数药物浓度-时间曲线先后出现快速下降的分布相和缓慢下降的消除相。
1.2 统计矩模型统计矩模型以概率论和统计矩理论解析血药浓度-时间数据,忽略房室的概念,因而也称非房室模型。其特征参数包括零阶矩、一阶矩和二阶矩[10]。零阶矩为血药浓度-时间的曲线下面积(AUC),是血药浓度和时间的综合参数,与给药量成正比;一阶矩为药物的平均滞留时间(MRT),反映消除速度;二阶矩为MRT的方差(VRT),反映药物分子在体内平均停留时间的差异。平均滞留时间MRT=AUMC/AUC(AUMC为Ct-t的曲线下面积),清除率CL=FX0/AUC(X0为给药量,F为生物利用度),MRT=t1/2/0.693,t1/2=0.693MRT,Vd=CL·MRT。认为药-时曲线末端的药物在体内分布平衡,仅有消除相,因此根据药-时曲线末端的斜率(2.303/K)计算药物的消除半衰期(t1/2=0.693/K)。统计矩模型的优点是限制性条件少,只要求药-时曲线末端符合指数消除,可以解决不能用房室模型拟合的问题。
2 模型的缺陷和不足
房室模型和统计矩模型能帮助人们理解药物在体内的分布和消除情况[1],但与人体的实际情况仍有距离。
2.1 房室模型房室模型历史悠久,是药代动力学的标准方法,但在应用过程中,由于测量误差,分布相与消除相的分界点有时难以判定,同一组数据很可能由于分界点的不同,计算出的药动学参数可能不同。同一个药物也会因采样点设计、给药方式的不同,很可能表现出不同的房室数目。当有两个以上模型与数据吻合时,很难确定哪个正确,使得房室模型拟合具有不确定性[11]。房室模型的一些参数比较抽象,如K12、K21,由于中央室和外周室的具体器官并不确定,因而无法用实验进行验证。中央室的分布容积(Vc),也因其器官的不确定,很难理解。
更为重要的是房室模型的理论有明显缺陷,它将人体的组织器官按药物的分布速率假设成不同的房室。一室模型认为,药物进入机体后,迅速随血流分布到各器官达到平衡,即药物分布到各器官的速度相同。二室模型认为,药物先随血流分布到血流速度大的器官,再向血流速度小的周边室分布。然而,人体各器官的血流速度是固有的,应该与药物的关系不大,一般不会因为药物的入血而明显改变器官的血流速度。
表1显示了一些主要动脉的血流速度。从表中的数据可知腹主动脉的血流到达各脏器的速度。供应中央室器官(脑、肝、肾)动脉的血流速度也很快,每分钟达几十米。以往研究认为,血流速度较慢的外周室的器官(如脂肪、肌肉)动脉的血流速度其实并不慢。脂肪主要分布在腹腔,主要由肠系膜动脉供血而肌肉主要在四肢。供应腹腔脂肪和肌肉的动脉血流速度并不明显低于脑、肝、肾,都能在几秒钟内到达各组织。单位时间内的血流量是血流速度的另一种表述单位。每搏心输出量约70 mL,每分钟心输出量约5 L[12]。根据主动脉的内径[13]可以估算出主动脉的容积为200~300 mL。也就是说,心脏每分钟的搏出量可以把主动脉的血更换20次,即很快能到达各脏器。因而,由于血流速度差异而导致不同房室的解释难以成立。
2.2 统计矩模型统计矩模型弥补了房室模型的缺点,限制性条件少,只要求药-时曲线末端符合指数消除,解决了不能用房室模型拟合的问题。统计矩模型公认的缺点是只能提供总体参数,不能提供血药浓度-时间的细节[16]。而且统计矩涉及的卷积分、逆卷积分等原理,普通的医药学研究者很难掌握其理论基础,在药动学参数测定中仅教条式地进行数据输入计算,甚至直接用软件进行结果输出,而对结果的内涵理解不深。
表1 主要动脉的血流速度[14]
Tab.1 Blood flow velocity of some arteries
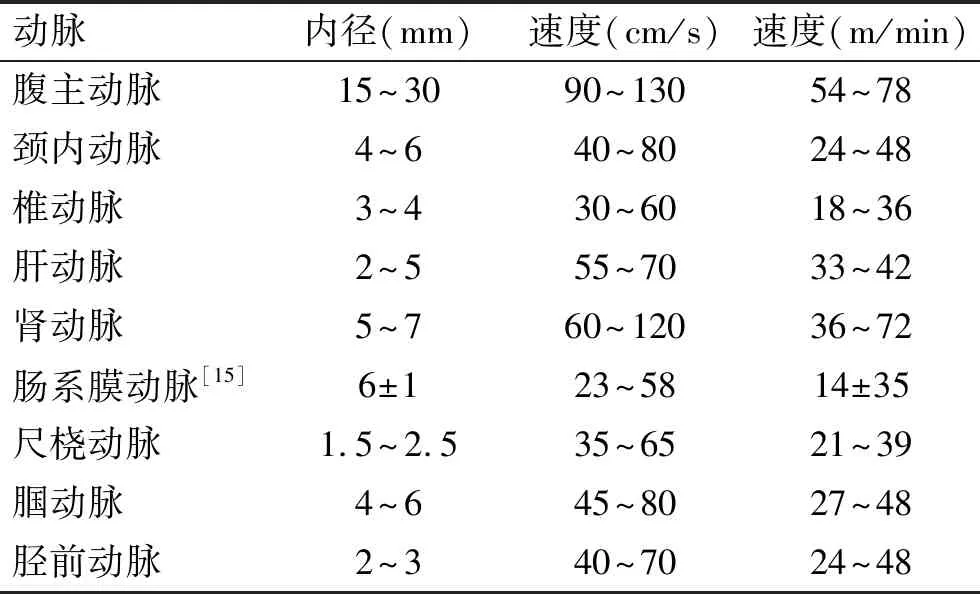
动脉内径(mm)速度(cm/s)速度(m/min)腹主动脉15~3090~13054~78颈内动脉4~640~8024~48椎动脉3~430~6018~36肝动脉2~555~7033~42肾动脉5~760~12036~72肠系膜动脉[15]6±123~5814±35尺桡动脉1.5~2.535~6521~39腘动脉 4~645~8027~48胫前动脉 2~340~7024~48
其实,统计矩模型的缺陷并不止此。其特征参数的零阶矩AUC是血药浓度随时间变化的指标,其本质是血药浓度,而不是体内药量。药物代谢动力学研究药物在体内的过程[17],而血药浓度不等于体内药量。当药物在体内分布平衡时,血药浓度与体内药物量的比值基本恒定,血药浓度的变化能反映体内药量的变化[18]。也就是说,药物在体内分布平衡时,血药浓度的变化率等于体内药量的变化率。统计矩模型没有强调药物的分布平衡,因而其血药浓度变化率不一定等于体内药量变化率。其所计算出的参数反映的是血药浓度的参数,而不是体内药量的参数。对于一室模式的药物,给药后体内药物迅速达到平衡,统计矩模型计算出的参数反映的是体内药量的变化。而对于二室或多室模型药物,计算出的参数并不反映体内药量的变化,而大多数的药物是二室模型。换句话说,统计矩模型仅适用于一室模型药物的分析,对于大多数(二室或多室模型)药物,统计矩模型的结果反映的是药物代谢动力学的表象,而不是本质。
统计矩模型根据曲线末端血药浓度计算药物消除半衰期[16],其弊端也显而易见:①曲线末端仅是整个消除相的一部分,由此推论整个消除相,以点代面存在风险;②末端相血药浓度低,距离检测限近,测量的误差大,势必导致测定的半衰期误差大;③药物的消除有一定的阈值,末端的血药浓度很可能接近或低于阈值,因而不一定符合消除相的规律;④末端相血药浓度较低,如果低于最低有效血药浓度,其临床意义值得商榷。
统计矩模型认为,MRT=t1/2/0.693[10,19]。然而,我们统计了2017年中国知网发表的统计矩模型(非房室模型)的论文,大多数情况下二者并不相等。说明MRT和t1/2/0.693单个或两个均有问题。因此,统计矩模型在药代动力学研究中正在不恰当地应用,甚至被滥用。应该引起关注。
3 改进方案
药物的体内消除是逐渐进行的连续过程。血管外给药后,药物的吸收、分布和消除几乎同时进行,静脉注射给药后分布和消除并存。吸收和(或)分布影响对消除相的研究。药物吸收完成、分布平衡后的药-时曲线反映药物的消除,据此可以分析药物消除的速率过程。药物消除的速率过程归纳为一级、零级和混合动力学3种类型。房室模型和统计矩模型武断地假设药物按一级动力学消除,根据假设进行计算,但最后并没有对假设做进一步验证,其结果的正确性值得怀疑。如果能找到一个仅有消除相的给药方法,就能简单、清楚地阐明消除相的规律,再依次弄清分布相和吸收相规律。用仅有消除相的药-时曲线,能直接判断药物的消除类型,简单方便地计算消除参数。
3.1 单纯消除相测定药物代谢动力学参数众所周知,任何给药途径连续多次给药5个半衰期后,血药浓度可以基本达到稳定状态,即稳态浓度。那么,缓慢恒速静脉滴注5个半衰期后,血药浓度能达稳态,同时药物在全身分布平衡,虽然各组织器官的药物浓度并不相同,但其比值基本恒定。此时停止给药,血药浓度的变化与体内药量的变化一致,即血药浓度的变化可反映体内药量的变化。在这种药物分布平衡的条件下,停药后不同时间采血,绘制的血药浓度-时间曲线的变化仅反映药物的消除,曲线的类型反映药物的消除类型。如果是一级动力学消除,可根据其消除规律(lnC=lnCss-Kt),将对数血药浓度与时间直线回归,得出消除速率常数(K)和停药前的血药浓度(Css),再计算出消除半衰期(t1/2=0.693/K)。静脉滴注达稳态(平衡)后体内药物总量Ass=1.44·Vt1/2(V为恒速静脉滴注速度,单位mg/h)。稳态时的血药浓度(Css)也可以直接测得,药物的表观分布容积(Vd)可以依据公式(Vd=Ass/Css)计算,进而计算出药物清除率(CL=K·Vd)。
3.2 恒速静脉滴注期间血药浓度测定药物代谢动力学参数恒速静脉滴注是等剂量等间隔多次静脉注射的极限状态,表现为恒定的给药速率和持续的零间隔。缓慢恒速静脉滴注一般在几个小时内将药量缓慢输入体内,体内各组织器官与血药浓度差明显小于静脉注射时的差。因而静脉滴注过程中,体内的药物分布基本是平衡的。也就是说,血药浓度与体内药量的比值基本是恒定的,血药浓度可以代替体内药量进行药代动力学分析。持续静脉滴注时,进入体内药物的速度是恒定的。大多数药物按一级动力学消除,即单位时间内消除恒定比例的药物,所以,药物消除量随血药浓度的升高而增加。静脉滴注后,血药浓度逐渐上升,当消除速度增高至静脉滴注速度时,血药浓度不再上升,达到稳定状态[17]。体内药量(A)等于血药浓度乘表观分布容积,即A=C·Vd。
恒速静滴过程中,体内药量(At)与时间的关系为:
At=A0+(Ass-A0)·(1-e-Kt)
则血药浓度与时间的关系可表示为:
Ct=C0+(Css-C0)·(1-e-Kt)[20]
式中Ct为t时间时的血药浓度,Css为稳态血药浓度,C0为药物消除的阈浓度,K为药物消除速率常数。Css、C0和K是常数,Ct和t呈指数关系。将静脉滴注期间不同时间点的Ct与时间t进行一阶指数回归,可计算出C0、Css和K。根据消除速率常数K可计算出t1/2(t1/2= 0.693/K)。
静脉滴注达稳态后体内药物总量Ass=1.44·V·t1/2(V为滴注速度,单位mg/h)[21]。稳态时的血药浓度(Css)也可以直接测得。药物的表观分布容积可以依据公式Vd=Ass/Css计算。进而,计算出清除率CL=K·Vd。
3.3 分布相参数的测定静脉注射给药时,药物先集中在血管。血管内的药物有血浆蛋白结合型和游离型。由于毛细血管内皮的连接有较大间隙,游离型药物可进入组织间液,然后再跨膜转运进入细胞内。血浆蛋白结合率高的药物很少进入外周组织,主要滞留血管,因而很快分布至全身;跨膜转运快的药物也能很快分布到全身的组织,这些药物的分布主要表现为一室模型。血液中的药物部分与血浆蛋白结合,游离型药物多经滤过进入组织间液,再经简单扩散等方式转运到细胞内。由于药物跨膜进入细胞的速度明显小于从血液经毛细血管滤过入组织液的速度,因而表现为二室模型。由于药物进入不同组织细胞的速度可能不同,或者细胞内的药物再进入细胞器,而表现为多室模型。
静脉注射给药的药-时曲线的药物分布特征明显。研究药物分布的动力学参数时,可测定静脉注射药物后不同时间的血药浓度,在药-时曲线的分布相上,减去消除相的药物浓度部分,得到单纯的分布相的血药浓度,再根据其曲线类型,计算分布相的药动学参数。
4 小 结
房室模型和统计矩模型是研究药物体内过程的基础和重要理论,是药物代谢动力学发展的基石。用其测定药动学参数的主要障碍是分布相的干扰,恒速静脉滴注期间和停药后体内药物的分布基本平衡,用其血药浓度-时间曲线测定药动学参数避免了分布相的干扰,我们称这种方法为分布平衡法或静脉滴注法。这种方法的完善和改进还需要大量的工作,但其必定会促进药代动力学的发展。
