瞬时受体电位M4通道与心脏电活动及心律失常的研究进展*
2019-08-31尹琳黄从新
尹琳 黄从新
瞬时受体电位M4(transient receptor potential melastatin 4 ,TRPM4)是一种在心脏中广泛表达的、与多种心血管疾病相关的阳离子通道。TRPM4具有胞内钙离子和电压门控双重依赖性,但与许多其他TRP通道不同,其仅对单价阳离子是可通透性的。多项研究发现TRPM4是心脏电活动的重要参与者,现笔者对此做一综述。
1 TRPM4通道的结构
最近有科学家相继揭示人类TRPM4蛋白的原子结构:TRPM4是由四个相同亚基围成的离子通道,其中每个亚基包含多个跨膜和胞质结构域,大致分为三个区域:N端为TRP通道家族的同源区域(MHRs),由MHR1~4组成;C端(CTD)有两个螺旋区域;跨膜区域(TMD)包括S1~S6区和TRP区(图1)。
2017年12月Winkler等[1]通过低温电子显微镜揭示了TRPM4通道的“王冠样结构”——C端的其中一个卷曲螺旋区域(coiled-coil domain)组成王冠的“顶”,而另一螺旋结构类似于王冠的“骨架”穿过MHRs,并与MHRs和TMD共同形成王冠的“帽状”(图2为平行于细胞膜平面的三维重建结构,四个亚基分别用四种不同的颜色标记)。几乎同一时间Autzen及其团队[2]利用脂质体包含钙离子结合的人类TRPM4和钙离子未结合的人类TRPM4,再用低温电子显微镜对这两者结构上的不同进行分析,揭示了TRP区和S1~S4结构域的基础结构,确定了S1~S4存在Ca2+的结合位点。他们提出当Ca2+结合至TRPM4通道时使得第905位点的精氨酸向位于第790位点的脯氨酸移动,造成了TRPM4通道的闭合状态与Ca2+未结合时的状态不同,即钙结合诱导构象变化,从而使得TRPM4通道具有电压依赖性。

图1人类TRPM4单个亚基的平面简图(来源于doi:10.1038/nature24674)
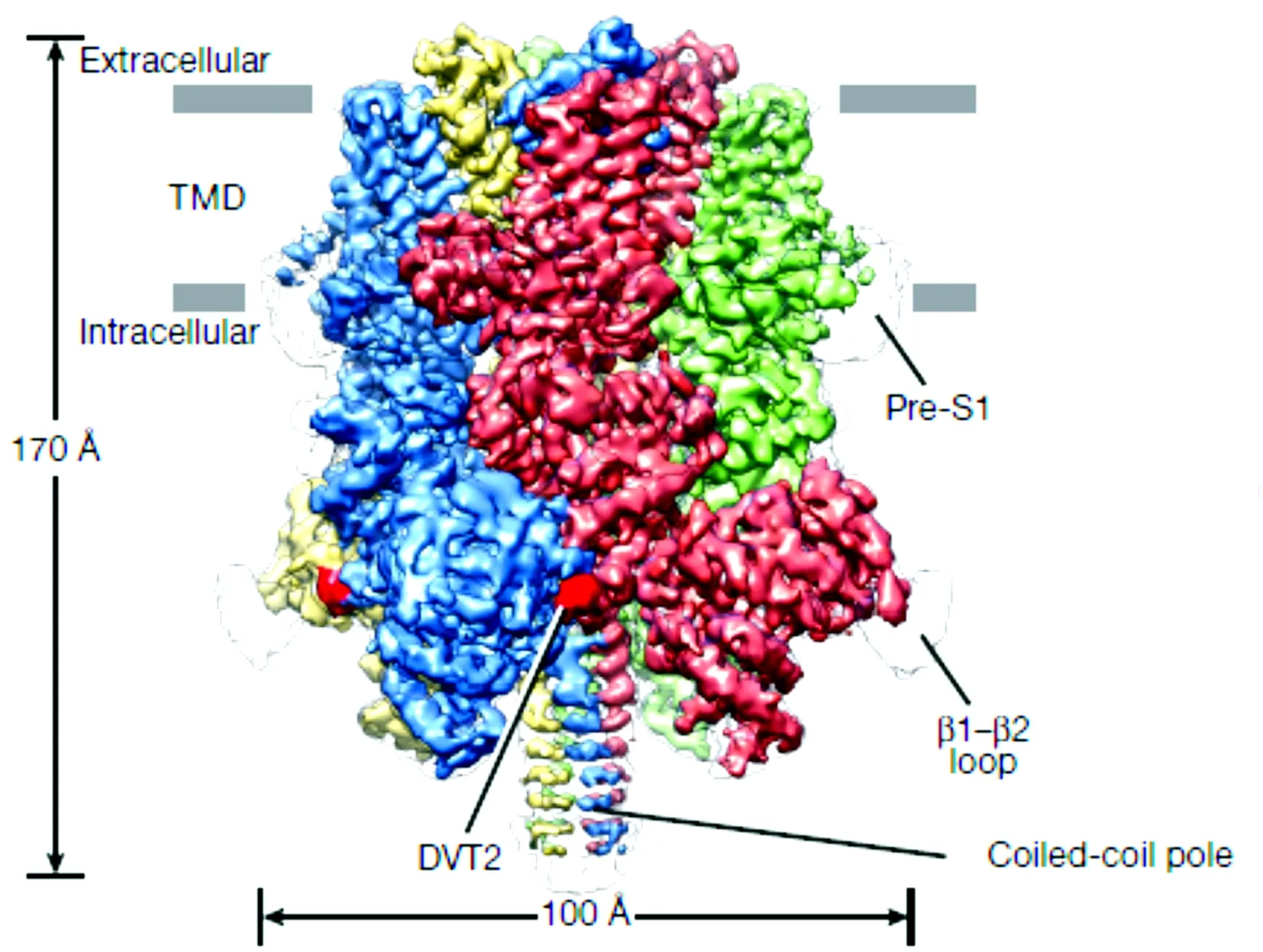
图2人类TRPM4的三维结构重建图 (来源于doi:10.1038/nature24674)
除了上述hTRPM4的结构外,guo等[3]采用相同的方式揭露了小鼠的TRPM4在ATP结合及未结合状态下的原子结构,发现其亚基的分区及整体的四聚体结构大致与人类的相似,只是对各区域的命名有少许差异,其将mTRPM4整个蛋白大致分为3层。底层结构位于胞质区,主要包含N末端核苷酸结合结构域(NBD)、锚定蛋白重复序列(ARD)和C末端卷曲螺旋。中层是连接子螺旋区域(LHD),包含12个连接子(LH1-LH12),形成了亚基内多个域间相互作用的支架。S1-S6六个跨膜区域和TRP区域形成亚基的最顶层,与其他的大多数经典的六个跨膜四聚体阳离子通道一样,可将其分为两个部分:S1-S4的电压感应域(VSD)和由S4-S5连接子螺旋连接的S5-S6孔结构域。
2 TRPM4通道在心脏中的表达
心脏是TRPM4表达的主要组织之一。近来一些研究发现TRPM4 mRNA或蛋白质表达有种属差异性:在正常人类心房当中可检测到TRPM4的蛋白和电流[4],通过对人类心脏中的TRPM4的mRNA的评估发现其主要表达于浦肯野纤维,而在心室肌细胞中表达量很少[5];牛心中的TRPM4蛋白在室间隔的心内膜表达量较心房、心室高[6]。在大鼠心脏中,TRPM4的mRNA、蛋白和电流主要出现于病理性心室肌当中,如低氧再灌注损伤[7]和自发性高血压模型[8]。小鼠的窦房结细胞也高表达TRPM4的mRNA和蛋白,其次是心房,心室表达量最少[9]。在功能层面上,TRPM4电流已在啮齿动物[10]以及哺乳动物[11]心肌细胞记录到。除了心肌细胞外,TRPM4的mRNA还存在于大鼠心室成纤维细胞[12]和血管平滑肌细胞[13]中。
3 TRPM4与心脏电活动
3.1TRPM4与窦房结 窦性心律是心脏电活动的起点,来源于窦房结细胞产生的自发的舒张期去极化( diastolic depolarization,DD)。DD是由超极化激活的环核苷酸门控通道(hyperpolarization-activated cyclic nucleotide-gated ion channel,HCN)[14]和局部钙释放激活的内向电流[15]所介导的。急性分离的小鼠窦房结细胞中存在功能性的TRPM4单通道离子流[9],且TRPM4基因突变的病人表现为心动过缓[16],因此TRPM4被认为是局部钙释放激活的内向电流的一个候选通道。
在急性分离的小鼠右房中,包含有窦房结,能产生自发的动作电位(action potential,AP),TRPM4抑制剂9-菲酚能减慢心率,然而在TRPM4敲除的动物模型当中却不能产生效应,说明9-菲酚是通过抑制TRPM4而不是其他离子通道起作用[17]。另外,在兔的窦房结中,9-菲酚能降低DD的斜率。但上述实验均是在离体的心房所进行,它失去了内源性神经-激素的影响,而且分离的心房肌的搏动频率比在体的心率慢(300次/分 vs 500次/分),所以并不能真正意义上说明TRPM4对窦房结的影响。Demion等[10]将12周至32周鼠龄的雄性小鼠随机分为两组:TRPM4基因敲除组(Trpm4-/-组)与对照组(Trpm4+/+组),予以12 h的夜间心电图(ECG)监测表明:Trpm4-/-组比Trpm4+/+组的窦性停搏的发生的次数多[(1.41±0.3)次, (n=18) vs (0.33±0.2)次,(n=13) ;P<0.05],这说明了TRPM4对维持稳定的在体窦性心律有一定的作用。窦房结中精确的局部钙离子释放水平现在还未知,计算机仿真实验表明1 umol/L的钙离子浓度变化将足够激活TRPM4,并且激活的TRPM4电流能够增加窦房结细胞的DD斜率[18]。
更有趣的是,9-菲酚的效应和内源性心率成负相关[17]:内源性心率越低,9-菲酚减慢心率的效果越明显,这说明了TRPM4仅仅在心脏节律降低的时候被激活,从而起到拮抗心动过缓的作用。此也能进一步解释为什么Mathar等[19]通过基因重组的方式将第一代(R1)大鼠中胚胎干细胞的TRPM4基因与 Cre-loxP-重组产生TRPM4无效等位基因,再经过反向杂交得到纯合子(全身性TRPM4基因敲除)于第八代(R8)用于实验,ECG却发现全身性基因敲除大鼠的心率与正常组的心率无明显差异[(524.6±10.3)次/分,(n=21) vs (502.5±2.6)次/分,(n=19);P<0.05]。另外,Kecskes等[20]的研究也印证了上述观点,该研究ECG示心脏特异性敲除TRPM4(TRPM4CKO)的小鼠与正常小鼠之间RR间期无明显统计学意义[(99.9±1.6)ms,(n=18) vs (97.5±0.9)ms,(n=13);P= 0.24]。为了获得TRPM4CKO小鼠,其团队将Trpm4L3F2小鼠与EIIa-Cre小鼠杂交。选择后代Trpm4flox小鼠进行进一步繁殖。随后,将Trpm4flox小鼠与MLC2a-Cre小鼠杂交,这导致TRPM4基因第15位、16位的外显子其编码TRPM4蛋白的第一跨膜结构域的心脏特异性缺失,进而成功产生TRPM4CKO小鼠(其中Trpm4L3F2小鼠为经过基因工程处理后带有特异性片段的小鼠;Trpm4flox小鼠为Trpm4L3F2小鼠与EIIa-Cre小鼠杂交,其后代中带有某种特殊位点的小鼠;基因片段详情请参考具体文献)。
由于人类的心率约为60~100次/分,所以相对于小鼠而言,9-菲酚的效果可能在人类心脏中较大。例如TRPM4的Y790H位点突变[16]能引起心动过缓,但遗憾的是关于该突变型的电生理学性质未有进一步研究,且关于TRPM4突变病人的系统评价[21]也没有提供TRPM4与心率相关的数据,这可能是缺少大数据研究病例造成的。
3.2TRPM4与工作心肌细胞 TRPM4也参与心房、心室肌AP的形成。Hu等[22]的研究表明:在离体的心房肌细胞(HL-1细胞)和心室肌细胞(Luo-Rudy心室肌细胞)中,TRPM4是钙离子和电压双重依赖性的,仅仅数倍(3~5倍)的TRPM4激活就足够延长AP复极化时程,进一步增加TRPM4的电流密度(≥6倍)能引起早期后除极(EADs)的叠加,从而产生一个EADs样的刺激易化去极化。这与血管紧张素Ⅱ作用于该两种细胞的效应相同,且这两者产生的效应均能被9-菲酚所逆转。在小鼠的TRPM4敲除模型当中,异丙肾上腺素能增加其心率[19],但在该动物模型分离的右房当中肾上腺素对其心率的影响不大[17],说明在体的肾上腺素刺激引起的心率增加可能不是直接通过调节TRPM4的活动所产生的结果。TRPM4对心肌AP的作用仍未完全明确,上述各个研究结果的差异性,可能是由于小鼠品系、取材部位或者检验手段不同引起。
现在关于TRPM4在心肌AP中扮演的角色主要概括为三点[23-24]:①TRPM4通道开放能加快去极化,因此出现在动作电位的起始部分;②TRPM4是被钙离子所激活,所以在AP过程中能进一步增强;③TRPM4对Na+和K+的通透性是同等的,推测在膜电位为负的时候,其为Na+通透性的内向电流,参与去极化,而在膜电位为正的时候其转为K+通透性的外向电流。因此,TRPM4激活使膜电位趋向零,这也是心肌AP平台期的膜电位。目前关于TRPM4对工作心肌细胞作用的研究局限于小鼠,而人类工作心肌细胞平台期(约占AP的1/2)[25]相对于小鼠(约占AP的1/3)来说占据AP的大部分时程,因此关于TRPM4在人类心肌细胞AP中扮演的角色值得深入探究。
4 TRPM4突变引起的心律失常
4.1传导阻滞 TRPM4突变能引起人类心脏传导性疾病最早被发现是在一个携带有常染色体显性遗传基因的束支传导阻滞的家庭当中,其中E7K突变被确定为TRPM4的突变位点[5],紧接着其他的突变位点相继被发现,如Stallmeyer等[16]通过对160位无遗传关系的心律失常的病人的基因型分析确定的Q131H、A432T、G582S、Y790H、K914R以及liu等[6]在法国和黎巴嫩一些家族中发现的R164W、A432T与G844D。这些突变位点大约占右束支阻滞的1/4、占房室传导阻滞的1/10。Liu表示TRPM4结构中的N端和胞内的环形结构的功能突变表现为进行性地传导阻滞,与心脏猝死有关,能引起心脏浦肯野纤维的变性。
上述提到的Demion等[10]的研究发现Trpm4-/-组小鼠表现出了多水平的传导阻滞,包括一度和二度I型房室传导阻滞。Trpm4-/-组小鼠二度Ⅱ型房室传导阻滞中PR间期进行性延长往往伴随着心率变异性短期评价指标——正常相邻RR间期差异性的平方根(RMSSD)的增加,这表明二度房室传导阻滞可能是由于发作性的副交感神经超速驱动所引起。为了进一步证实该猜测,该团队采取阿托品干预Trpm4-/-组与Trpm4+/+组,经过6 h的体表心电图记录发现阿托品确实能减少Trpm4-/-组的6 h内二度房室传导阻滞的发生频率[Trpm4-/-+阿托品组(0.6±0.3)次 vs Trpm4-/-组(5.0±0.4) 次,P<0.01],对Trpm4+/+组无明显影响[Trpm4+/++阿托品组(1.4±0.3)次 vs Trpm4+/+组(1.5±0.2)次]。相对的是,在阿托品作用下,Trpm4-/-组与Trpm4+/+组的平均PR间期未见明显改变[(42.3±0.3 )ms vs (42.3±0.3 ) ms,P<0.001],说明一度房室传导阻滞不是由于副交感神经过度激活所引起,可能将归因于离子通道的突变或者结构的改变。
Liu等[6]发现了R164W、A432T与G844D这三种TRPM4突变后,对这几种突变进行了深入的研究:该团队采用II型快速定点诱变试剂盒作用于TRPM4的cDNA,从而产生了TRPM4Arg164Trp、TRPM4Ala432Thr、TRPM4Gly844Asp、正常组(WT组)四种类型的质粒,再利用这四种质粒分别去转染中国仓鼠的卵巢细胞(Chinese hamster ovary,CHO)、人胚胎肾细胞和非洲绿猴细胞(COS-7)这三种细胞,最后应用膜片钳技术记录各组的电流与导电率,其实验主要集中于人胚胎肾细胞。实验结果显示在人胚胎肾细胞中三种突变类型的电流幅度[TRPM4Arg164Trp( 91.1±20.9)pA/pF,n=5 vs TRPM4Ala432Thr(86.3±21.0)pA/pF,n=5 vs TRPM4Gly844Asp(152.6±15.7)pA/pF,n=5]均比WT组高[(18.8±3.7 )pA/pF,n=10],但均没有引起单个通道的导电率和对钙离子的亲和力的改变(具体数值文献未给出)。过表达动力蛋白(dynamitin)是破坏动力蛋白运动系统和干扰离子通道内吞作用的既定方法[26]。紧接着,该团队采用dynamitin分别和上述四种类型的质粒共转染人胚胎肾细胞,发现突变的TRPM4通道电流对dynamitin不敏感[TRPM4Arg164Trp(120.0±24.9 )pA/pF,n=5、 TRPM4Ala432Thr(83.6±23.0)pA/pF,n=5 、 TRPM4Gly844Asp(153.9±11.2)pA/pF,n=6],而WT组的电流密度增加[(42.7±8.7 )PA / PF,n=7]。由此得出结论:TRPM4突变损害了基于动力蛋白的内吞作用。SUMO蛋白修饰分为三步,而Ubc9是第二步的关键调节酶,影响着真核细胞中SUMO与其靶蛋白的结合,SENP1是去SUMO化蛋白修饰的调节剂。为了进一步明确该胞吞作用可能涉及到的转录后修饰,他们研究了Ubc9和SENP1对TRPM4电流密度的影响。和上述方法一样,他们应用Ubc9、SENP1分别与四组质粒共转染HRK-293细胞。WT组与Ubc9的共表达使得TRPM4电流密度显著增加[(43.3±12.7)pA / pF,n=9,P=0.04],WT 组与SENP1的共表达使TRPM4电流密度[(4.5±1.0) pA / pF,n=8]降低至背景电流水平[(3.5±0.4) pA / pF,n=7,P=0.39],而三组突变型的细胞与Ubc9 共表达[(63.3±7.1) pA / pF,n=17,P=0.46]或SENP1[(64.5±17.2) pA / pF,n= 6,P=0.65]对电流密度均无明显影响。这些结果意味着去SUMO化充当TRPM4内化的信号,因此,SUMO的蛋白修饰保护突变的TRPM4通道蛋白免于内吞作用和蛋白酶体降解,从而引起细胞膜TRPM4通道蛋白的表达密度增加。
4.2Brugada综合征(BrS) 部分BrS病人是由于TRPM4通道突变引起的[16, 21, 27]。Liu等[21]对248名BrS患者(排除了SCN5A突变)的TRPM4基因序列进行了分析,在20个相互独立的个体中发现了11种TRPM4的突变位点,这些发现表明TRPM4突变占BrS的约6%。他们选择了其中的4种(P779R、T873I、L1075P和K914X)突变用来进一步的电生理学研究,为了得到突变体,先将野生型的TRPM4的cDNA克隆至pcDNA4 / TO载体,再用定点诱导试剂盒进行体外诱变。深入的研究表明:K914X位点突变导致TRPM4蛋白的截短从而形成无功能的离子通道;另一个突变,P779R,相对于野生型TRPM4来说,其降低了全身的TRPM4电流,但没有引起单价阳离子通透性的变化;T873I和L1075P位点突变相对正常组而言,在TRPM4电流记录上没有显著的统计学差异。这种差异性说明了TRPM4功能表达异常虽然能引起BrS,但各个突变位点产生的效果却各不相同。Durhoit等[27]和Stallmeyer等[16]均只是对临床BrS患者进行遗传学分析,发现TRPM4突变是BrS的一种候选基因之一,但未进行进一步的电生理探究。
上述的突变分析表明TRPM4电流密度的增加或减少均能引起传导阻滞,有学者认为这可能是通过降低Nav1.5钠通道的活性而形成的[28]:功能的增加可以使膜的静息电位去极化并因此使钠通道失活,而功能的丧失可以导致膜电位的超极化进而降低细胞的兴奋性和传导。这些假定的作用机制可以部分解释SCN5A功能丧失和TRPM4突变两种不同病变有某些相似表型(例如均表现为BrS)这一现象。
5 TRPM4与心脏肥大引起的心律失常
尽管对于心脏肥大引起心律失常的机制还未完全明确,但Ca2+似乎是这一现象出现的主要组成部分[29]。细胞内Ca2+浓度升高时内向电流的激活可诱发心律失常,至今发现的三种胞内Ca2+浓度敏感的内向电流包括:钠钙交换体、Ca2+激活的氯化物电流和Ca2+激活的非选择性阳离子电流[30]。延迟后除极(delayed after depolarization, DADs)发生在AP的4期,是在复极完成后发生的短暂震荡性除极活动,为瞬时内向电流(Iti)所引起,主要为Na+。但DADs发生的主要机制是细胞内Ca2+浓度增加,肌浆网的钙负荷过重,即形成钙超载,使细胞膜对Na+通透性增加,因此只要能引起细胞内Ca2+超负荷的因素均可以促发这一离子流。有Guinamard等[8]利用自发性高血压(SHR)所致的心肌肥厚大鼠模型和膜片钳技术发现TRPM4的离子特性与Iti相符,均受胞内钙离子浓度的调控,该研究也发现TRPM4在高血压大鼠心室肌中的表达高于正常的Wistar大鼠。因此认为TRPM4可能参与钙超载的形成,进而促进DADs。
在心肌AP的形成过程中TRPM4参与致心律失常的EADs形成,延长APD。有数项证据支持这一结论:①EADs和TRPM4的激活均与肌浆网的钙释放相关,TRPM4的激活电压在心肌细胞AP去极化发生过程的范围内,因此TRPM4的激活可使得EADs持续性产生[31];②研究表明心肌肥厚能引起EADs[32];③在SHR动物模型中钙瞬变的发生率增加,其能促进钙敏感性的离子通道电流的增加从而调整APD[33],而TRPM4在SHR中也能延长APD[34]。
6 结语
总的来说,上述数据已经表明TRPM4与心脏电生理有联系,我们目前还未完全明确。尽管关于TRPM4突变和遗传性心脏疾病的关联已得到初步的认识,但仍有很多问题待解决:首先,研究者已在HEK-293细胞中初步证实了某些突变能修饰TRPM4蛋白和电流,但其有待在心脏细胞中进一步得到验证;其次,过表达或者敲除TRPM4动物模型的建立可以为其对心脏电活动的影响提供部分机制学说,但关于TRPM4在心脏中扮演的角色还未完全得到认识,尤其是在心脏传导阻滞中;最后,随着TRPM4敲除小鼠模型的建立,TRPM4在心脏发育和心血管系统中的作用仍需深入研究。
最近对于TRPM4结构的进一步解析对理解其功能有很大的帮助。另外,细胞水平的电生理学实验能深入探究TRPM4与膜电位的关系。因此,笔者认为TRPM4有可能成为抗心律失常药物的新靶点或者副作用的来源。
