Progress in molecular-simulation-based research on the effects of interface-induced fluid microstructures on flow resistance☆
2019-08-19YumengZhangYudanZhuAnranWangQingweiGaoYaoQinYaojiaChenXiaohuaLu
Yumeng Zhang,Yudan Zhu,*,Anran Wang,Qingwei Gao,2,Yao Qin,Yaojia Chen,Xiaohua Lu
1 College of Chemical Engineering,State Key Laboratory of Materials-oriented Chemical Engineering,Nanjing Tech University,Nanjing 211816,China
2 Energy Engineering,Division of Energy Science,Luleå University of Technology,Luleå 971 87,Sweden
Keywords:Process intensification Nanoconfined fluid Interface Complex fluids Micro structure Molecular simulation
A B S T R A C T In modern chemical engineering processes,solid interface involvement is the most important component of process intensification techniques,such as nanoporous membrane separation and heterogeneous catalysis.The fundamental mechanism underlying interfacial transport remains incompletely understood given the complexity of heterogeneous interfacial molecular interactions and the high nonideality of the fluid involved.Thus,understanding the effects of interface-induced fluid microstructures on flow resistance is the first step in further understanding interfacial transport.Molecular simulation has become an indispensable method for the investigation of fluid microstructure and flow resistance.Here,we reviewed the recent research progress of our group and the latest relevant works to elucidate the contribution of interface-induced fluid microstructures to flow resistance.We specifically focused on water,ionic aqueous solutions,and alcohol-water mixtures given the ubiquity of these fluid systems in modern chemical engineering processes. We discussed the effects of the interfaceinduced hydrogen bond networks of water molecules,the ionic hydration of ionic aqueous solutions,and the spatial distributions of alcohol and alcohol-water mixtures on flow resistance on the basis of the distinctive characteristics of different fluid systems.
1.Introduction
Several bottleneck problems,such as environmental pollution,resource shortage,and energy overconsumption,are encountered in process industries[1-3].New-generation chemical engineering separation and reaction technologies,which are represented by membrane separation and heterogeneous catalysis,are breakthrough technologies that are expected to solve these bottleneck problems[4,5].In modern chemical engineering,process intensificationis mainlyachieved throughinterfaceintroduction.Forexample,in nanoporous membrane technology(e.g.nanofiltration and reverse osmosis),the difference in interactions between nanopore walls and different fluid molecules is exploited to enable separation without phase transition and greatly reduce energy consumption[6,7].Our limited knowledge of the mechanism underlying the mass transfer of fluid molecules confined within the nanospaces of membrane materials leads to the permeabilityselectivity trade-off[8].Similarly,in the heterogeneous catalysis process,the constant increase in the surface areas of catalysts results in the nanoconfinement of reactants and products.However,the transport behaviors of nanoconfined molecules to and from catalytic sites remain incompletely understood[9-12].Therefore,the interfacial mass transfer of nanoconfined fluid molecules is a ubiquitous process in modern chemical engineering and a fundamental scientific problem.
Table 1 presents a summary of the main differences between traditional and modern chemical engineering process.The focus of modern chemical engineering processes has drastically shifted from traditional ideal,isotropic bulk fluids(e.g.hydrocarbons)to highly non-ideal,anisotropic nanoconfined fluids (e.g. ionic aqueous solutions and alcoholwater mixtures)because of the continually increasing requirements of the energy and environmental fields[7].
The use of classical macroscopic mass transfer models[13,14]to describe the interfacial transport behavior of nanoconfined fluids remains challenging because these models fail to consider the effect of the interface on fluid molecules.Lu et al.proposed an interfacial mass transfer model based on linear non-equilibrium thermodynamics to provide a universal framework for nanoscale interfacial transfer.The mass transfer rate is equal to the ratio of the driving force to resistance,as shown in Eq.(1)[15].

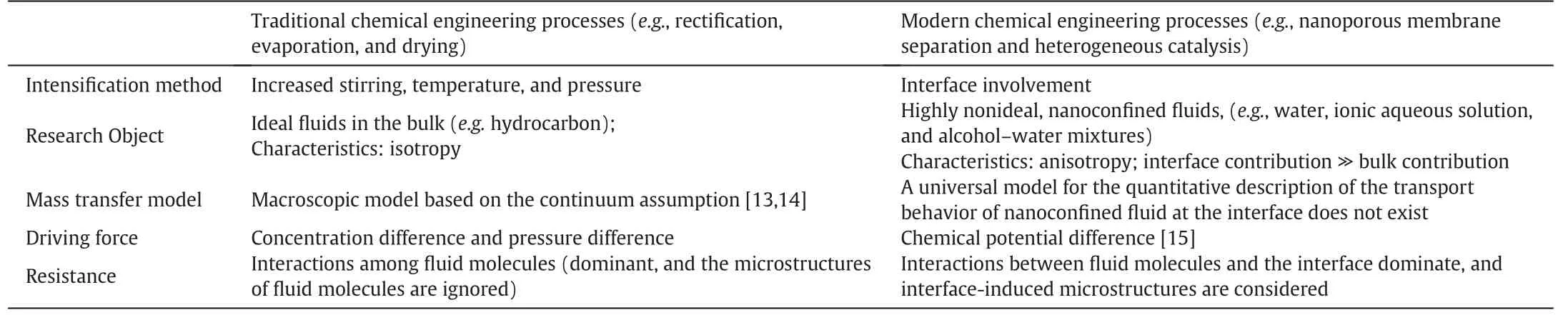
Table 1Main differences between traditional and modern chemical engineering processes
where μ0represents the chemical potential at system equilibrium;μ-μ0represents the chemical potential difference,which is the driving force;and 1/k represents mass transfer resistance.This interfacial model has been applied in the analyses of the rate-controlling step in the dissolution of K2SO4crystals in different solvents[16-18],the ion-exchange rate of the synthesis of K4TiO4whiskers[19-21],and the separation of CO2by supported ionic liquid (IL) membranes [22,23]. This model,which is based on chemical potential,could facilitate the qualitative analysis of resistance in interface involvement.
The development of a predictive model based on Eq.(1)first requires a full understanding of the effect of the interface on nanoconfined fluid behavior.As shown in Table 1,the interactions between fluid molecules and the interface dominate on the nanoscale.Developments in experimental characterization and molecular simulation techniques have provided considerable evidence for the close association between the interface-induced microstructures of fluid molecules and anomalous flow behavior on the nanoscale[24-29].Thus,we reviewed the recent research progress of our group and the latest relevant works to understand the effects of interface-induced fluid microstructures on flow resistance.Understanding the interfacial mass transfer of nanoconfined fluids is the first step toward explaining anomalous phenomena and determining the governing factors that enhance process intensification in modern chemical engineering.
Molecular simulation has become an indispensable and primary investigative method in modern chemical engineering,given the lack of nanoscale in situ experimental techniques.Gubbins et al.summarized theories and modeling methods at different length and time scales(including electronic,atomistic,mesoscale,and continuum level methods)[30].Electronic level methods(quantum mechanics)focuses more on the distribution of electron cloud structure around a single molecule or atom,instead of the behavior of molecules.In mesoscale methods(e.g.dissipative particle dynamics and coarse graining),a model particle represents an entire molecule or a fragment containing multiple molecules without considering the details of the atomic behavior.Continuum methods can well reflect the flowing of the macroscopic bulk fluid,but they are not applicable when describing at the nanoscale.Molecular simulation at the atomistic level of description is a suitable method to study interfacial mass transfer processes.Given that the microstructures of fluid molecules often have a great influence on the resistance of fluid flowing,the molecular simulations method involved in this paper focused on at the atomic level.Palmer et al.reviewed the development and challenges of molecular simulation from a chemical engineering perspective[31].We have performed extensive systematic studies based on molecular simulations to investigate the relationship between flow resistance and nanoconfined fluid microstructures.Molecular simulation provides diverse parameters for the characterization of fluid flow resistance,such as friction coefficient,residence time,mean square displacement(MSD),and diffusion coefficient.
In this review,we focused on the flow behavior of three typical and common fluid systems in modern chemical engineering process:water,ionic aqueous solutions, and alcohol-water mixtures (Fig. 1). We reviewed the recent research progress of our group and the latest relevant works to elucidate the contribution of interface-induced fluid microstructures to the flow resistance of nanoconfined fluid.We summarized the effects of interface-induced hydrogen bond (HB) networks in water molecules,the ionic hydration of ionic aqueous solutions,and the spatial distributions of alcohol-water mixtures on the basis of the distinctive characteristics of each fluid system.
2.Effects of the Interface-induced HB Networks of Water Molecules on Flow Resistance
The behaviors of water molecules nanoconfined on interfaces have become popular topics in modern chemical engineering research because water is the most commonly used solvent in industrial production and is widely used as a medium in numerous chemical reactions[32].The microstructures of nanoconfined water molecules play an important role in fluid flow[33-35].

Fig.1.Research object and content.
HB structure is among the most important indices that reflect the microstructures of water molecules [36].The HB structures of water molecules are defined by two criteria:the geometric criterion applied by Mezei et al. [37,38] and the energetic criterion implemented by Rahman and Stillinger[39,40].Each water molecule in the bulk phase exhibits a tetrahedral structure with approximately 3.5 HBs[41-45].Advanced experimental techniques, such as nuclear magnetic resonance(NMR),time-resolved infrared spectroscopy,X-ray absorption spectroscopy,and X-ray Raman scattering can provide valuable information on local HBs;the measurements obtained through these techniques, however, may present experimental errors given that these techniques require the introduction of other substances into the experimental system[23,25,43,46].Molecular simulation complements experimental methods because it can enable single-factor investigations.Currently, molecular simulation is widely used to characterize HB properties,such as the average number,distribution,and HB lifetime[41,47-49].
The original HB tetrahedral network of water molecules could be changed by the properties of the interface.The following two subsections present a review of the different effects of the interface-induced HB networks of water molecules on flow resistance in accordance with different confinement dimensions.
2.1.One-dimensional nanotubes
Our research group has performed numerous molecular simulation studies on one-dimensional(1D)carbon nanotubes(CNTs),and systematically evaluated the effects of size[50],helicity[50,51],surface properties[52,53],temperature[53]and external electric field[54]on the behavior of water molecules confined in CNTs.Zhu et al.summarized relevant works and pointed out that the influence of the modified group of the pore interface on water molecule microstructure is far greater than that of the external conditions(e.g.,temperature and electric field)[55].
The mobility of water is closely associated with the variation in the structure of HBs under nanoconfinement.Wang et al.[50]found that the average number of HBs decreased from 3.49 to 1.37 when water molecules were confined in hydrophobic CNTs with diameters of 0.676 nm.Moreover,water molecules formed unique single-file water chains when confined in CNTs with diameters of 0.676-0.811 nm;these water chains are an important cause that the flux of water molecules in CNTs is of comparable magnitude to that in aquaporin-1[24].Single-file water chains have also been successfully observed through in situ Raman spectroscopy in 2010[56].On the other hand,the diffusion coefficient can reflect the resistance of fluid flowing.Nanoconfined fluids transport mainly in the form of diffusion,sometimes controlling the rate of the whole process. The diffusion coefficient D represents the diffusion flux under the unit concentration gradient, which expresses the diffusion speed of a component in the medium.The atomic diffusion coefficient has a close connection with the amount of MSD.The definitions of MSD and diffusion coefficient in 1D nanotubes are shown in Eq.(2)and Eq.(3),respectively[57].Cao et al.studied the microstructures and dynamic properties of water molecules confined inside and outside CNTs and titanium dioxide nanotubes(TiNTs)with diameters of 1.0 nm. They found that the diffusion coefficients of water molecules confined in TiNTs were lower than those of water molecules in the bulk phase,whereas those of water molecules confined outside CNTs exceeded those of water molecules in the bulk phase(2.49×10-9m2·s-1).The difference in diffusion coefficients may be attributed to the different HB microstructures of water molecules that formed within tubes with different interfacial properties[47].These results demonstrated that the diffusion coefficient of confined water is closely associated with the interface-induced HB structure. Furthermore, different interfacial properties would greatly affect the flow resistance of water molecules by causing variations in the HB structure of water molecules under nanoconfinement.

2.2.Two-dimensional nanoslits
Since the discoverers of graphene won the Nobel Prize in 2010,various two-dimensional(2D)materials have received considerable attention[58-63].Consequently,the behavior of water molecules confined in 2D nanochannels has become a popular research topic[8,64,65].The HB structures of water molecules confined in 2D slits are distinct from those of water molecules confined in 1D nanotubes.Prisk et al.found that water molecules confined in zinc silicate hemimorphite could form a 2D HB network(i.e.in-plane structure)that deviated from the tetrahedral structures of water molecules in the bulk phase and could enhance the fast rotational diffusion of water molecules[66].Our findings on 1D nanotubes posed the following questions for subsequent research:(I)What parameters can improve the characterization of water molecules under 2D nanoconfinement?(II)What is the relationship between the change in HB structure and the flow resistance of water molecules? (III) What is the importance of this relationship in the applications of nanoporous materials?
Residence time is an effective indicator of the flow resistance of water molecules nanoconfined at the interface[67,68].Wei et al.performed molecular dynamics simulations to study the structure and diffusion of water confined in rutile TiO2(110) and graphite (0001)nanoslits with widths of 0.8 nm to 2.0 nm at room temperature.Their results showed that the residence time of water molecules on graphite surfaces was 0.5%of that of water molecules on TiO2surfaces.Comparing the HB microstructures of water molecules on these surfaces revealed that interfacial properties influenced HB formation, which affected water molecule diffusion.Similar to the 1D confinement,the diffusion coefficient of water molecules in hydrophobic graphite was greater than that of water molecules in hydrophilic TiO2.The diffusion coefficient is mainly influenced by the formation of HBs between water molecules and the interface.The less HBs formed by water molecules results in the faster diffusion.Therefore,the effect of surface chemistry on flow resistance is greater than that of surface curvature[69,70].
The use of most TiO2-supported catalysts in heterogeneous reactions(e.g.hydrodesulfurization)requires elevated temperatures.Molecular dynamics simulation was applied to investigate the temperature-dependent structural properties of water molecules confined in rutile TiO2(110)nanoslits.The mean residence time of the first layer of water molecules decreased dramatically at temperatures above 300°C,which indicated that water molecules had escaped from the TiO2surface.The layer-bylayer analysis of HBs revealed that the formation of HBs within the first layer of water molecules was largely responsible for the reduction in the mean residence time and that the generation of 1D HB networks enhanced the removal of water molecules,as shown in Fig.2[71].Similar findings were found for TiO2surfaces with other crystal planes.Bahramian et al.studied the temperature-dependent effect of water on anatase(001)and(101)surfaces at 273-373 K.They found that waterwater interactions drastically intensified with increasing temperature.This behavior indicated that reductions in water-TiO2interactions promoted the escape of water molecules from the surface[72].
These simulation findings provided the following insights on the interfacial modification of mesoporous TiO2for further practical applications: (I) Given the difficulty in removing the water layer from the TiO2surface,organic molecules with strong competitive adsorptive ability are a better choice than water molecules for the modification of TiO2surfaces.(II)In reactions with polar products,the strong interactions between TiO2and the product molecules may decelerate the reaction rate of the products.Therefore,interfacial modification is required to reduce the flow resistance of products at the catalyst interface[71].On the basis of the above guidelines,Li et al.proposed a novel method for the modification of the interfacial properties of a mesoporous TiO2-supported catalyst for hydrodesulfurization.Their proposed method involved grafting with phenylphosphonic acid,which has stronger competitive adsorption ability than water. After carbonizing the catalyst surface,the heterogeneous surface modification of carbon considerably improved the molecular transport ability of the polar product molecules of H2S at the catalyst interface and facilitated H2S removal from the catalyst interface.The final desulfurization conversion rate of the modified catalyst increased from 65%to 98%relative to that of the unmodified catalyst[73].Molecular simulation can provide useful insights on the applications of nanoporous materials.

Fig.2.(a)Mean residence time of water molecules of different layers on TiO2 surface as a function of temperature.(b)Number of water molecules with i(i=1,···,4)H-bond per unit area of the TiO2 surface(n1,···,n4)[71].Copyright 2016 Elsevier B.V.
In addition to residence time, the friction coefficient between the fluid molecules and the solid interface is another important parameter for the evaluation of fluid flow resistance.Zhu et al.proposed the simulation method of nanofriction-based nonequilibrium molecular dynamics (NEMD) for the investigation of the interfacial flow resistance of fluid molecules.In contrast to traditional NEMD methods,which initiate fluid motion by exerting driving force on fluid molecules, the nanofriction-based NEMD method facilitates the directional motion of fluid molecules by moving two pore walls in opposite directions.This method is more convenient than other methods that require the use of experimental nanoscale force measurement apparatus (e.g. atomic force microscope[AFM]).Nanofriction-based NEMD has been used to investigate the effect of wall properties on the flow resistance of water molecules confined between two Si(111)surfaces. Results indicated that the flow resistance of confined water molecules was contributed by(I)the interactions between water molecules and the wall and(II)the interactions among water molecules themselves.Flow resistance increased as hydrophilicity increased.The variations in the orientation of the first layer of water molecules near the interface caused by the intensifying interfacial hydrophilic effect increased the probability of the formation of HBs among interlayer water molecules and further enhanced interactions among water molecules[74].
The flow resistance of water molecules confined within TiO2interfaces was analyzed [75]. The velocity profiles of all water molecules nanoconfined in the first layer and those of some water molecules confined in the second layer were consistent with those of the TiO2walls during friction.This observation was consistent with the mobility characteristics of water molecules on the interfaces of TiO2particles identified through NMR[76].The study also found that the variation in interface-induced HB properties depended on the the friction coefficient. As slit width increased, the friction coefficient eventually decreased because the lifetime of HBs formed within and beyond the first layer of water molecules decreased,whereas the corresponding number of free water increased.This finding expanded our understanding of the molecular-scale differences between the friction coefficients of mesoporous and dense TiO2films observed through AFM experiments[77].Thus the mechanism underlying the reduction in the friction coefficient with the increasing roughness of the TiO2surface was revealed.
To summarize,pore size shape(1D and 2D confinement)and pore wall interfacial properties are important factors that influence HB microstructure.The interface-induced HB microstructure is highly dependent on the flow resistance of nanoconfined water molecules.
3.Effects of the Interface-induced Ionic Hydration of Ionic Aqueous Solutions on Flow Resistance
Ionic aqueous solutions play a critical role in the processes of solid interface involvement in modern chemical engineering.Some of these processes include the desalination of sea water and the recovery of lithium from lake brine through membrane separation and the development of battery systems.Accordingly,relevant theoretical research is crucial for determining the influence of the solid interface on the microstructure of ionic aqueous solutions. Ionic hydration is the essential characteristic of ionic aqueous solutions and cannot be ignored when studying the flow behavior of ions under interfacial influence[29,38,78].When the electrostatic interaction between water molecules and ions is stronger than the HB interaction between water molecules,the original tetrahedral HB structure of water molecules is destroyed.The destruction of the original HB structure,in turn,changes the orientation of water molecules.Thus,instead of traveling alone,an ion in aqueous solutions is tightly surrounded by a layer of water molecules during transport.This water shell,which is usually referred to as the ionic hydration microstructure,can be treated as a unitary water sphere.
The high nonideality of ionic aqueous solutions can be mainly attributed to the ionic hydration microstructure and resulting asymmetric molecular interactions.In accordance with the calculated radial distribution function between the ion and water molecule,the definition of the ionic hydration microstructure includes ions and water molecules in the first ionic coordination shell.This unified definition, however,cannot fully reflect the differences among different types of ions,especially ions under nanoconfinement by a solid interface. Extensive molecular simulation investigations[79-88]have indicated that the definition of the ionic hydration microstructures of different types of ions varies.Fig.3 illustrates that not all water molecules in the first coordination shell of ions with weak hydration ability are hydrated.By contrast,water molecules in the first and second coordination shells of ions with strong hydration ability are hydrated.
To improve our understanding of the mechanism underlying ion separation and explain anomalous phenomena that occur on the nanoscale,we summarized a series of molecular simulation studies that investigated different ionic characteristics to determine how to(I)reasonably quantify the ionic hydration microstructure of different ion types,(II)determine the effect of interfacial properties on the ionic hydration microstructure,and(III)identify the relationship between interface-induced ionic hydration and the flow resistance of ionic aqueous solutions.
3.1.Ions with weak hydration ability(e.g.K+)
Zhou et al.proposed a new concept of hydration factor(F)to determine the effective hydration microstructures of ions with weak hydration ability in accordance with the orientation of water molecules in the first coordination shell(Fig.3a)and the hydration factor for cations and anions are defined in Eq.(4)and Eq.(5),respectively[85].A low F value corresponds to weak hydration ability(e.g.,the value of F for bulk K+is only 0.56[84]).F can well reflect the different effects of the interface on the order of hydration shells of different ions.A low F value indicates that the ionic hydration microstructure is vulnerable to damage when the interface is introduced. Shao et al. found that the change in F was dependent on the interaction energy between the ion in the hydrophobic pore and its surrounding water molecules. This quantitative dependence relationship is crucial for explaining K+/Na+separation.Therefore,the definition of microstructures with close associations with energy is necessary to define different ionic hydration microstructures[55].

To further analyze the hydration factor,Zhu et al.studied the different responses of K+and Na+to the transport resistance exerted by carbonyl groups modified on the internal walls of CNTs.They found that the effect of carbonyl groups on the walls varied with the order of water molecules in the first coordination shells of different ions.The order of water molecules in the first coordination shell of K+was more sensitive to the modified groups of CNTs than in the first coordination shell of Na+.In addition,the chemical modification of CNTs with different sizes could exert contradictory effects on the ionic hydration microstructure.For example,the electrostatic effect dominated among CNTs with large pore sizes and high hydration order.By contrast,the steric hindrance effect dominated among CNTs with small pore sizes and low hydration order. These results illustrated the different responses of the modified group to different hydrated ions on the molecular scale[87].Guo et al.applied the F value to explain the special affinity of 18-crown-6 for K+.By analyzing the F values obtained under bulk and nanoconfined conditions,they found that nanoconfinement simultaneously strengthened the hydration of K+and Na+and that K+was more sensitive to nanoconfinement[81].Furthermore,the sensitivity of K+to the electric field effect induced by wall charge was stronger than that of Na+.The electric field exerted the dominant effect among large pore sizes, whereas confinement exerted the dominant effect among small pore sizes[80].In summary,the analysis of the influence of nanoconfinement, surface properties,and electric field effect on F showed that the hydration microstructures of different ions have different sensitivities to these factors and that the hydration microstructure of K+is more sensitive than that of Na+.

Fig.3.Schematic of ionic hydration microstructure[85,88].Copyright 2002 Elsevier B.V;Copyright 2017 American Chemical Society.
On the basis of F-value analysis and previous findings,Zhu et al.proposed a novel strategy for the regulation of ionic flow resistance.Their proposed strategy was based on an electric field and could be used to regulate separation.Their analysis of 2D density distribution revealed that K+tended to localize in the center of CNTs because of its weak interaction with modified groups(low resistance).Na+,however,tended to present a circular distribution between the modified group and the center of the CNT because of its strong interaction with the modified group(high resistance). Differences in the distribution patterns of K+and Na+were closely associated with the characteristics of their ionic hydration microstructures.As electric field intensity increased,carbonyl groups tilted toward the direction of the electric field. This behavior increased the cross-sectional area of the modified region.This effect,in turn,reduced ionic transport resistance by increasing the probability of K+and Na+to localize in the central region.The resistances of K+and Na+,however,decreased at different rates because their interface-induced ionic hydration microstructures exhibited different responses to the electric field.As electric field intensity increased,the resistance of K+decreased faster than that of Na+.This behavior enhanced K+/Na+selectivity[86].
3.2.Ions with strong hydration ability(e.g.Mg2+and Li+)
The definition of the ionic hydration microstructure of ions with strong hydration ability should not solely depend on the characteristic of the first coordination shell. The method presented in Fig. 3(b)shows that the multilayer of water molecules surrounding the ions represents an integral ionic hydration microstructure.Water molecules surrounding ions could be divided into different regions in accordance with their spatial distributions.Thus,HBs should be fully considered in addition to ionic hydration.
Mg2+and Li+are typical cations with strong hydration ability.These cations are difficult to separate because they have similar atomic radii and their first coordination shells have similar F values of close to 1.The difference between the ionic transport resistances of Mg2+and Li+in nanopores must be studied because the membrane separation of these ions is essential for extracting lithium from lake brine[89,90].Ruan et al.[82]found that Mg2+/Li+could be separated by using membranes with the optimal pore size range of 0.76-1.04 nm,which coincides with the sizes of biological channel filters selective for Mg2+(CorA[91]and GMN motifs[92]).They then illustrated the separation mechanism of these ions by analyzing their ionic hydration microstructures with close associations with energy.They found that Mg2+/Li+separation was mainly dependent on structural differences between the second ionic hydration shells of Mg2+and Li+.The results of 2D density distribution analysis showed that Mg2+tended to localize in graphene pores(low resistance).This distribution pattern would prevent Li+ions from approaching the graphene pore(high resistance)through ionic blocking.Profiling the mean force potential of the ions indicated that the energy barrier encountered by Li+when moving through pores was larger than that encountered by Mg2+.Furthermore,they analyzed the orientation distributions and the HB structures of water molecules surrounding ions per layer. They found that when ions traveled through nanopores,the interface-induced ionic hydration of the different ions varied.This variation,in turn,led to variations in the orientation of water molecules surrounding different ions and resulted in the formation of different HB structures.Moreover,compared with the second hydration shell of Mg2+,that of Li+required the removal of more water molecules and the breakage of more HBs.This requirement increased the resistance encountered by Li+when moving through pores and promoted the separation of Mg2+/Li+.Thus,this requirement essentially accounted for the different pathways followed by Mg2+and Li+through pores and the energy barrier that the ions need to overcome(Fig.4)[82].

Fig.4.(Left)(a)selectivity filter structure of CorA Mg2+channels viewed from the top.(b)Graphene nanopores with pore diameters of 0.789,(c)1.024,and(d)1.501 nm.(e)Lateral view of the simulation box showing a graphene nanopore in the center and the two reservoirs on both sides of the nanopore.The black arrow represents the direction of the applied electric field.The gray rectangle frame is the simulation box.(Right)Average H-bond numbers per water molecule resulting from water molecule formation along the z-axis in the first and second hydration shells(type I;a and b)and hydrogen bonds formed in water molecules in the second hydration shell(type II;c and d).The insets illustrate hydrogen bond formation in the ionic first and second hydration shells.The blue dashed line between water molecules represents H-bonds[82].Copyright 2016 American Chemical Society.
Zhu et al.designed porous graphene with a five-coordinate group that conformed to the characteristics of selective filters in biological Mg2+channels and investigated the effect of different types of groups(i.e.--CO and--COO-)on ionic hydration microstructure and flow resistance.They found that increasing the average interaction energy difference between Mg2+and Li+increased the flux and selectivity of--COO--modified nanopores. In contrast to those of Mg2+and Li+under confinement by pristine porous graphene[82],the dehydration numbers of the second hydration shells of Mg2+and Li+under confinement by different modified nanopores were almost the same.Therefore,to distinguish the effect of the modified groups on the hydration microstructures of different ions,they considered the F value of water molecules in the second hydration shell(F2).Their results indicated that Mg2+and Li+selectivity was mainly affected by F2 values.Different F2 values could reflect the different orientations of water molecules in the second hydration shell.Different orientations resulted in different HB structures and further affected transport resistance.To further investigate the interactions between water molecules, they classified HBs into Hb-1 and Hb-2. Hb-1 was formed through the interaction among water molecules between hydration shells,whereas Hb-2 was formed through the interaction between water molecules in the first hydration shell and the water molecules and oxygen atoms of the modified group in the second hydration shell. The Hb-2 of nanoconfined Mg2+was almost consistent with its bulk counterpart. The oxygen atoms in the modified group(i.e.--CO and--COO-)could perfectly replace the water molecules in the second hydration shell of Mg2+and thus reduce the transport resistance of Mg2+,whereas the second hydration shell of Li+continued to dehydrate and break HBs.Hence,Hb-2 is an essential index for the analysis of the effect of different modified groups on ionic hydration microstructures.The separation of Mg2+/Li+is facilitated by the presence of oxygen atoms in the modified group;these oxygen atoms can perfectly replace water molecules in the second hydration shell of the ions(Fig.5)[88].
3.3.Complex ions with multiple atoms
The variation in the interface-induced microstructures of complex ions with multiple atoms has received increased attention with the rapid development of supported IL membranes and IL-immobilized sorbents for gas separation[93-96].In solution,however,complex ions with multiple atoms (e.g., IL and deep eutectic solvents) have more complex molecular interactions than ions with single atoms.These interactions include not only ionic hydration but also ion-pairing [79,97]. Changes in external conditions (temperature [98,99], pH [100],water content[79,101])could alter microstructure composition in solution, and may sometimes be dominated by ionic hydration or ion pairing.The hydration shells of ions with multiple atoms must first be defined. The definition of the hydration shells of ions with multiple atoms,however,is more difficult than that of ions with single atoms.
Choline chloride/urea(ChCl/urea)is a deep eutectic solvent that is widely used because of its low price and considerable potential for large-scale industrial applications.Gao et al.proposed that the hydration microstructure of each atom should be defined on the basis of their hydration ability and the HB network of surrounding water molecules (Fig. 3c) [79]. Using this approach, they defined the hydration structure of choline ions.Their simulation results showed that choline and chloride ions were not fully hydrated even when the water content reached 90%.This phenomenon suggested that ionic hydration and ion pairing always compete. Ion pairing was the dominant factor that influenced the microstructure of aqueous solutions with low water content.By contrast,ionic hydration dominated as water content increased.Furthermore,the competition between ionic hydration and ion pairing resulted in some highly non-ideal macroscopic experimental phenomena.Xie et al.found that excess molar volumes(VE)initially decreased and then increased with increasing water content.The molecular mechanism underlying this phenomenon could be elucidated through molecular simulation with reasonably defined fluid molecule microstructures[102].
The microstructural changes exhibited by complex ions will become increasingly complex when an interface is introduced to the bulk phase.These changes affect the mobility of complex ions and the transport resistance of gas molecules.Shi et al.studied the effect of temperatures of 313,423,and 573 K on the resistance of ILs([hmim][Tf2N]) and gas molecules (CO2, H2, N2, and H2O) confined in silica slit pores. They found that the diffusion coefficient of the IL decreased with increasing temperature. Moreover, the diffusion coefficient of gas molecules in the confining ILs was several times that of gas molecules in bulk ILs[99].These findings indicated that the influence of the interface was greater than that of temperature and provided guidance for the application of ILs in CO2capture.
Findings for nanoconfined ionic aqueous solutions demonstrated that the definition of interface-induced ionic hydration microstructures differ for different types of ions.The definition of a microstructure that is closely associated with energy based on ionic features is the key problem and the starting point in the exploration of flow resistance.Only the orientation of water molecules in the first coordination shell of ions with weak hydration ability should be considered.The hydration and HBs of ions with strong hydration abilitiy should be fully considered.The hydration and pairing of complex ions with multiple atoms should be considered.
4. Effects of the Interface-induced Spatial Distributions of Alcohol-Water Mixture on Flow Resistance
Alcohol is a potential alternative to fossil fuel.The flow behavior of alcohol molecules (methanol, ethanol, propanol, and butanol) in nanopores has crucial effects on interface involvement in modern chemical engineering processes.Alcohol produced through biological fermentation fails to meet industrial requirements given its low purity and high water content.Therefore,the separation of alcohol-water mixtures under membrane nanoconfinement is an essential process in modern chemical engineering [103-107]. Moreover, the analysis of fluid flow resistance is essential for the separation of alcohol-water mixtures.

Fig.5.(Left)Average number of H-bonds between the first and second ionic hydration shell per water molecule of the first ionic hydration shell along the z axis for(a)Mg2+and(b)Li+for co_5 and(c)Mg2+and(d)Li+for coo_5 under different electric field intensities(solid line,Hb-1 and dashed line,Hb-2).(Right)Average interaction energy per water molecule of the first ionic hydration shell at the entrance of the co_5 and coo_5 nanopores under different electric field intensities.The dashed lines indicate the bulk values for comparison[88].Copyright 2017 American Chemical Society.
Numerous experimental studies on the separation of alcohol-water mixtures by using various membranes, including organic, inorganic,and hybrid membrane materials,have been performed.Pore size and surface modification are among the most important factors that reduce flow resistance[108].Simultaneously increasing the selectivity and permeability of membranes,however,remains challenging because of the existing trade-off phenomenon[8].To overcome the trade-off phenomenon,the unusual microstructures exhibited by nanoconfined alcoholwater mixtures must first be understood. Molecular simulation will enable the investigation of the changes in interface-induced fluid microstructures and the determination of factors needed to guide related experiments.Molecular simulation studies have provided evidence that the interfacial-induced spatial distributions of alcohol (including molecular orientation and occupancy)under nanoconfinement have a considerable influence on the flow resistance of alcohol and alcoholwater mixtures[104,109-111].The unique spatial distributions of fluid molecules result in the formation of unique microstructures.
In reference to the above evidence,we reviewed the recent research progress of our group and the latest relevant works to address the following questions:(I)How do interfacial properties affect the microstructures of alcohol and alcohol-water mixtures? (II) What is the difference between the dominant factors that affect the flow resistance of pure alcohol molecules and alcohol-water mixtures?(III)What is the relationship between interface-induced spatial distributions and flow resistance?
4.1.Nanoconfined alcohol molecules
The contributions of influential factors(e.g.,pore size,surface geometry,and surface chemistry)on the flow resistance of alcohol molecules under nanoconfinement have been investigated.The relevant articles are listed in Table 2.Previous works have shown that the regulation of some influential factors could change the orientation distributions of alcohol molecules and thus change flow resistance and mobility.Ghoufi et al.evaluated the effects of surface properties(hydrophilicity and hydrophobicity)and pore size on the friction coefficient of tert-butanol confined in silica nanopores. They found that three layers of butanol molecules formed in hydrophilic pores(radius:1.2 nm)and that hydroxyl groups in butanol between layers were distributed in accordance with a specific orientation to form a stable cluster structure.Only two layers of butanol molecules formed in hydrophobic pores (radius:0.8 nm).The destruction of the original interlaminar structure reduced the friction coefficient of nanoconfined tert-butanol.The decrease in pore size prolonged dipole relaxation time in the confinement medium.Consequently,the flow resistance of tert-butanol increased,whereas its diffusion decreased[112].Moreover,the flow resistance of fluid molecules is mainly influenced by the difference in order degree caused by the change in orientation.Falk et al.investigated the flow resistance of ethanol confined in graphitic nanopores with different curvatures.The friction coefficient decreased as nanopore curvature increased because the order of the molecular orientation of ethanol increased [113].Shao et al. studied the structural properties of ethanol confined in CNTs with different diameters. Ethanol molecules confined in CNT(8,8)showed highly ordered structures,and most presented gauche configurations. As the degree of the order of ethanol molecules increased,their axial diffusion capacity decreased[109,114].Garberoglio investigated the effects of pore size and CNT flexibility on the flow resistance of methanol molecules.They found that the methanol microstructure was sensitive to pore size.Methanol molecules presented a chainlike structure in CNTs with small pore sizes(single file for CNT[6]and zigzag for CNT[8]).As pore size increased,the molecular orientation of methanol changed,and methanol molecules formed a helix structure.When pore size increased to 1.4 nm, confined methanol molecules tended to assume structures similar to those assumed by methanol molecules in the bulk phase.The variation in structure caused by molecular orientation affected flow resistance.The diffusion coefficient of methanol decreased as the diameters of the CNTs decreased.These phenomena indicated that the increased molecular structural order of methanol confined in hydrophobic CNT increased resistance.The effect of pore wall flexibility on flow resistance was similar to that of structural order.Flexible CNTs could improve the order of methanol molecules and thus reduce diffusion.The diffusion of methanol in rigid CNTs was approximately 10 times that in flexible CNTs[115].
Highly ordered fluid molecules may form a solid-like structure,which will affect flow resistance and exert different effects on the resistance of nanopores with different surface properties.Terrones et al.examined the structural properties and flow resistance of fluid molecules with different polarities(acetone,ethanol,methanol,N-methyl-2-pyrrolidone, carbon tetrachloride, and water) confined in hydrophobic graphene slit pores.Their results showed that all fluid molecules spontaneously formed ordered layers oriented parallel to the graphene surface.Moreover,the presence of a special 2D solid-like phase restricted the mobility of fluid molecules[116].Li et al.investigated the flow resistance of water, methanol, and decane (polarity: water >methanol>decane) confined in hydrophilic MoS2nanoslits with different slit widths(1.2,1.6,and 2.0 nm).The drastic differences in the friction coefficients of the three fluids indicated that polarity exerted the dominant effect on the flow resistance of nanoconfined fluid.The friction coefficients of the fluid systems increased as fluid polarity increased, i.e.water >methanol >decane. Moreover, the formation of a solid-likelayer enhanced the reduction in the resistance of the systems,and the ability of decane to form a solid-like layer was the strongest,followed by that of methanol and that of water(Fig.6)[111].

Table 2Summary of molecular simulation studies on the flow resistance of alcohol molecules

Fig.6.(Left)The friction coefficient of water,methanol and decane molecules confined in the MoS2 slits with different slit widths.The labels of“d12”,“d16”and“d20”indicate that the slit widths are 1.2 nm,1.6 nm and 2.0 nm,respectively.(Right)Density profiles along the Z-direction and X-Z direction snapshot of fluid molecules confined in a 1.2 nm MoS2 slit.(a)water(b)methanol(c)decane.Higher peak density means that molecules near the wall are more ordered[111].Copyright 2018 Elsevier B.V.
4.2.Nanoconfined alcohol-water mixtures
The effects of the molecular occupancy of alcohol-water mixtures on fluid flow resistance and separation performance are stronger than those of the molecular orientation of alcohol.Articles related to the molecular simulation of the influence of pore size,alcohol concentration,temperature,electric field,and other factors on fluid flow resistance under nanoconfinement are summarized in Table 3.
The annular layered structure of 1D-restricted pores promoted separation, and molecules that occupied the center of these nanopores showed low resistance. Furukawa et al. investigated the diffusion of ethanol-water mixtures with different compositions under confinement in a NaA zeolite crystal.They found that molecular occupation determined the magnitude of molecular resistance.When water molecules occupied the pore mouth,ethanol molecules had to overcome a large energy barrier to pass through nanopores. The diffusion coefficient of water,however,decreased as ethanol concentration increased[117].Lu et al.investigated the separation of ethanol-water mixtures with different mole fractions in the bulk phase and under confinement in zeolite and CNTs with different pore sizes.Under confinement in zeolite,water and ethanol molecules were randomly distributed,and water/ethanol was separated through screening.By contrast,the resistance of CNTs to water with ethanol mole fractions increased.CNT(7,7)was selective for the molar fraction ranges of ethanol. These phenomena could be attributed to the formation a single-file ethanol chain structure that occupied the central region of CNT(7,7)and exhibited reduced resistance.By contrast,water molecules formed an HB network that surrounded ethanol molecules and increased resistance[104].Wang et al.explored the effects of the oscillation period of the electric field on the transport properties of a methanol-water mixture(0.5 mol fraction)confined in CNT(12,12).The oscillation period of the electric field drastically influenced the flow rate of water and methanol molecules. In the absence of an electric field,methanol molecules preferentially occupied CNT pores.This behavior resulted in the low flow rate of water molecules.Under an external electric field,water molecules were more likely than alcohol molecules to occupy the surfaces of CNTs. This difference likely resulted in thedifference in resistance during the transfer of water and alcohol molecules to pores and resulted in their separation(Fig.7)[118].

Table 3Summary of molecular simulation literature on the flow resistance of alcohol-water mixtures
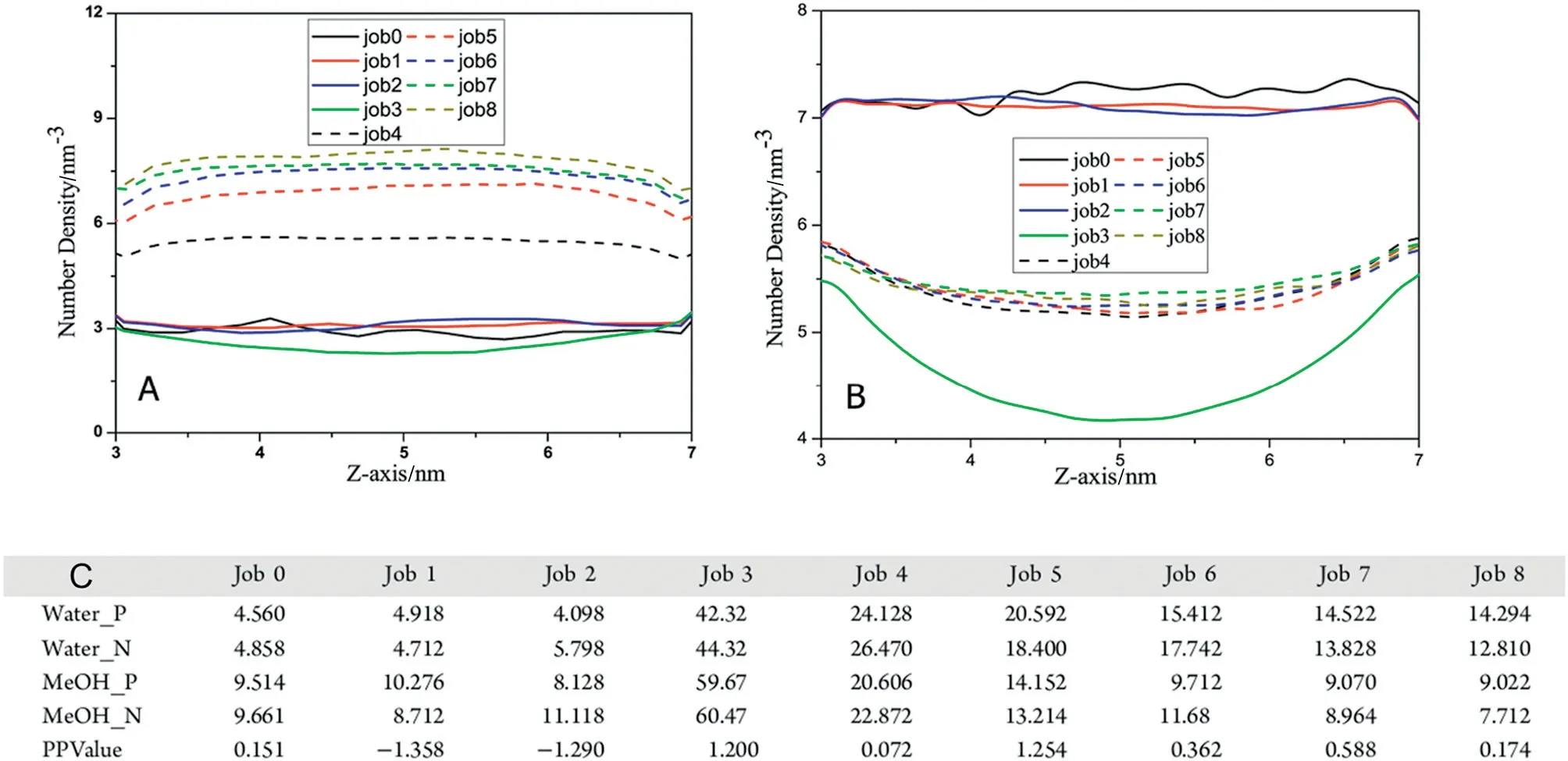
Fig.7.(A)Number density distributions of water molecules inside the CNT along the Z axis;Number density distributions of methanol molecules inside the CNT along the Z axis.(C)Flow rates of water and methanol molecules for each Job.Water_P(Water_N):The flow rate of water along the positive(negative)direction.MeOH_P(MeOH_N):The flow rate of methanol along the positive(negative)direction.PPValue=|Water_P-Water_N|-|MeOH_P-MeOH_N|.Job 0 represents the system without electric field;Job N(N=1,2…8)represent the systems with 0.5,1.0,5.0,20.0,40.0,60.0,80.0 and 100.0 ps oscillation periods[118].Copyright 2017 American Chemical Society.
Demixing in the direction parallel to the surfaces of 2D nanoslits is essential for examining resistance and separation. Dai et al. studied the effects of pore size and alcohol concentrations on the overall flow resistance of ethanol/water mixtures confined in graphene nanoslits.They found that the friction coefficient of the fluid system decreased as slit width increased.The addition of ethanol to pure water abruptly increased the friction coefficient of the fluid system.Therefore,interactions between ethanol molecules and the graphene surface are essential factors that influence the friction coefficient.Ethanol molecules preferentially adsorbed on the surfaces of large slits in graphene.This phenomenon resulted in demixing,which allowed interactions between ethanol molecules and the surface to remain unchanged. Therefore,the change in slit width and ethanol concentration negligibly affected the friction coefficient[119,120].Borges et al.studied the mechanism underlying the separation of water-alcohol mixtures(0.5 mol fraction)confined in multilayered GO membranes. The formation of a water monolayer on the GO surface promoted water diffusion.The rapid diffusion of water molecules simultaneously impeded the diffusion of alcohol molecules because of the demixing of alcohol and water molecules in the pores.This phenomenon enhanced selectivity because the membrane nanopores were almost entirely occupied by water molecules,whereas alcohol molecules were rejected[121].Different components of alcohol-water mixtures preferentially adsorb on different surfaces.The newly confined structures that form between a component(alcohol/water)and the interface would affect the transport resistance of the other component (water/alcohol) and separation performance.Water molecules are preferentially adsorbed on hydrophilic surfaces,and water/alcohol selectivity increases as the mole fraction of water molecules increases [122]. Alcohol molecules are preferentially adsorbed on hydrophobic surfaces.Gao et al.studied the behavior of several common alcohol-water mixtures confined in hydrophobic graphene slits [110].Under 2D nanoconfinement,alcohol molecules were preferentially adsorbed on the inner surfaces of hydrophobic graphene slits to form an alcohol adsorption layer,and water molecules were restricted between two alcohol adsorption layers.Moreover,different types of alcohol exhibited different demixing effects, which could change the residence time of water molecules on the alcohol layer(Fig.8). Hence,in alcohol/water separation, the formation of a confined structure between alcohol molecules and the surface will affect the transport of water molecules and alcohol/water separation.
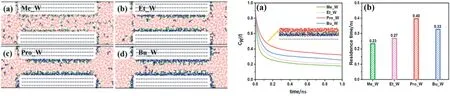
Fig.8.(Left)Snapshots of equilibrium configurations(side views)for(a)methanol/water,(b)ethanol/water,(c)propanol/water,and(d)butanol/water in contact with the graphene slit.(Right)Water molecules on the preferential adsorption layer of different alcohol molecules.(a)Residence autocorrelation as a function of time and(b)the residence time.The inset snapshots show the layered structure of propanol molecules and water molecules near the graphene wall[110].Copyright 2017 American Chemical Society.
Molecular orientation is the dominant factor that affects the flow resistance of pure alcohol molecules.Molecular occupancy is the dominant factor that influences fluid flow resistance and separation of alcohol-water mixtures.Moreover,interfacial properties greatly affect the spatial distribution of alcohol and alcohol-water mixtures,thus affecting the flow resistance of these fluid systems.
5.Conclusions
Interfacial fluid molecular transport is a ubiquitous phenomenon in modern chemical engineering processes,such as membrane separation and heterogeneous catalysis.It is also a bottleneck scientific problem because it involves highly complex heterogeneous interfacial molecular interactions and highly nonideal fluids.The effects of interface-induced fluid microstructures on flow resistance must first be unraveled to obtain additional insight on the fundamental mechanism of interfacial transport.In this review,we summarized the results of studies on the effects of interface-induced fluid microstructures on the flow resistance of water,ionic aqueous solutions,and alcohol-water mixtures,which are three typical and common fluid systems in modern chemical engineering processing.
Different nanoconfined fluids exhibit different dominant interfaceinduced microstructures.The interface-induced HB network is the most important factor that affects the flow resistance of water molecules.Notably,the flow resistance of water molecules could be reduced by the formation of some special structures, such as single-file chains and inplane structures,under confinement.The flow resistance of ionic aqueous solutions is mainly affected by interface-induced ionic hydration microstructures.The orientation distribution of water molecules around an ion must first be determined to define the ionic hydration microstructure,which may change the flow resistance of ionic aqueous solutions.The interface-induced spatial distribution of fluid molecules mainly influences the flow resistance of alcohol-water mixtures.The newly confined structures that form between a component(alcohol/water)and the interface would affect the transport resistance of the other component(water/alcohol)and separation performance.Thus,two fluids can be separated on the basis of the difference between their interfacial interactions.
In this review,we demonstrated the indispensability of molecular simulation as a method for the single-factor analysis of fluid microstructures and flow resistance.Molecular simulation studies have provided useful insight that can be applied to facilitate process intensification in modern chemical engineering.Advancements in molecular simulation and in situ characterization techniques will enable our full understanding of the interfacial transport mechanism in the near future.
Nomenclature
F hydration factor
F2 F value of water molecules in the second hydration shell
J mass transfer rate
VEexcess molar volumes
μ0chemical potential at system equilibrium
μ-μ0chemical potential difference
1/k mass transfer resistance
杂志排行

Chinese Journal of Chemical Engineering的其它文章
- Recent advances in acid-resistant zeolite T membranes for dehydration of organics☆
- Process intensification in vapor-liquid mass transfer:The state-of-the-art☆
- Extractive distillation:Advances in conceptual design,solvent selection,and separation strategies☆
- Beyond graphene oxides:Emerging 2D molecular sieve membranes for efficient separation☆
- A review of internally heat integrated distillation column☆
- Recent progress and future prospects of oil-absorbing materials☆