HPLC法测定利福布汀原料药及胶囊中有关物质Δ
2019-08-15易秋艳崔学文罗军刘文跃袁军
易秋艳,崔学文,罗军,刘文跃,袁军#
(1.四川省食品药品检验检测院微生物中心,成都611731;2.四川明欣药业有限责任公司质量部,成都 611130)
结核病是由结核杆菌感染引起的慢性传染病,肺结核对个人、家庭和社会的危害极大[1]。利福布汀为利福霉素S的螺旋哌啶衍生物,在临床中主要用于分支杆菌感染所致疾病如结核及鸟分枝杆菌-胞内分支杆菌复合体(MAC)感染,其对结核杆菌的抑制作用比利福平约强4倍[2],应用前景良好。目前,利福布汀(原料药)及利福布汀胶囊仅一家企业生产,批准文号为国药准字H20070294(利福布汀)、H20070296(利福布汀胶囊);现行质量标准为国家食品药品监督管理局批准的YBH05792007[3](利福布汀)、YBH05812007[4](利福布汀胶囊),在这两个标准中,有关物质的检查均采用高效液相色谱(HPLC)法测定,色谱系统相同,但笔者经试验后发现此现行方法中利福布汀与相邻杂质峰不能达到有效分离,杂质检出率较低,经网上检索后也未见利福布汀有关物质检查研究的相关文献报道。为此,笔者参照《美国药典》(USP)[5]、《欧洲药典》(EP)9.0[6]和相关文献[7-15],对现行方法进行了改进,以提高杂质检出率,提高对该产品的质量控制水平。
1 材料
1.1 仪器
LC-2010CHT高效液相色谱(HPLC)仪(日本Shimadzu公司);2695 HPLC仪(美国Waters公司);CPA225D十万分之一电子天平(德国Sartorius公司)。
1.2 药品与试剂
利福布汀原料药(批号:15070356、16050656、17020156,纯度:96.6%、97.3%、97.0%)、利福布汀胶囊(批号:16010157、16010257、17090257,规格:0.15 g)均来源于四川明欣药业有限责任公司;利福布汀对照品(中国食品药品检定研究院,批号:130586-200901,纯度:97.2%);水为超纯水,乙腈为色谱纯,磷酸二氢钾、氢氧化钠、盐酸等均为分析纯。
2 方法与结果
2.1 色谱条件与系统适用性试验
2.1.1 色谱条件 色谱柱:Agilent XDB-C8(250 mm×4.6 mm,5μm);流动相:乙腈-0.1 mol/L磷酸二氢钾溶液(用2 mol/L氢氧化钠调pH至6.5±0.1)(50∶50,V/V);流速:1.0 mL/min;检测波长:254 nm;柱温:30 ℃;进样量:20 μL。
2.1.2 系统适用性试验 取约10 mg利福布汀原料药,用2 mL甲醇溶解后,加2 mol/L氢氧化钠溶液1 mL,静置4 min,加2 mol/L盐酸溶液1 mL中和,再用流动相稀释至50 mL。取此溶液10 μL注入液相色谱仪,记录色谱图,利福布汀峰前应出现1个大的及2个小的降解产物峰。利福布汀峰与相邻降解产物峰之间的分离度应不小于1.3;理论板数按利福布汀峰计应不低于2 000。系统适用性试验色谱图见图1A。

图1 系统适用性试验高效液相色谱图Fig 1 HPLC chromatograms of system suitability tests
2.2 溶液的制备
2.2.1 供试品溶液 (1)原料药。取适量本品,精密称量,以流动相溶解并稀释制成每1 mL中含利福布汀1.0 mg的溶液,即得。(2)胶囊。取内容物适量,精密称量,以流动相溶解并稀释制成每1 mL中含利福布汀1.0 mg的溶液,滤过,取续滤液即得。
2.2.2 对照溶液 取适量利福布汀对照品,以流动相溶解并稀释制成每1 mL中含利福布汀0.01 mg的溶液,作为对照溶液。
2.2.3 灵敏度溶液 取对照溶液适量,以流动相准确稀释20倍,即得。
2.3 线性关系考察
取用流动相逐级稀释的利福布汀对照品系列溶液(每 1 mL 中含利福布汀 0.000 8、0.001、0.010、0.012、0.016 mg),测定峰面积,以质量浓度为横坐标(x)、峰面积为纵坐标(y),进行线性回归,得线性方程为y=4.114×107x+7.178×102(r=1.000 0),利福布汀的检测质量浓度线性范围为0.000 8~0.016 mg/mL。
2.4 专属性试验
取利福布汀原料药(批号:17020156)和胶囊(批号:16010157)内容物适量,分别加入2 mol/L盐酸溶液1 mL,破坏15 min,得酸破坏后样品;用0.25 mol/L氢氧化钠溶液2 mL,破坏2 min,得碱破坏后样品;用6%过氧化氢溶液2 mL,破坏8 min,得氧化破坏后样品;用130℃高温破坏6 h,得高温破坏后样品;用大于4 000 lx的光近距离照射7 d,得光照破坏后样品;取上述各破坏后样品及未破坏的样品按“2.2.1”项下方法制备成供试品溶液并进样检测。结果,检出多个降解峰,且均不干扰主峰的检测。专属性试验色谱图见图2。
2.5 检测限与定量限考察
确定测定峰高约为基线噪音3倍时为检测限、10倍时为定量限,结果利福布汀的检测限为0.025 4 μg/mL,定量限为0.085 2 μg/mL,定量限相当于供试品进样量的0.009%,灵敏度符合要求。
2.6 精密度试验
取“2.3”项下利福布汀对照品溶液(0.010 mg/mL),连续测定6次,计算得利福布汀峰面积的RSD为0.07%(n=6),表明仪器精密度较好。
2.7 重复性试验
分别取同一批利福布汀原料药及胶囊样品,按“2.2.1”项下方法制备成供试品溶液,平行6份,测定供试品溶液中利福布汀及其杂质总含量。结果,原料药(批号:17020156)中杂质总含量的RSD为1.8%(n=6),胶囊(批号:16010157)中杂质总含量的RSD为1.4%(n=6);原料药(批号:17020156)中利福布汀含量的RSD为0.77%(n=6),胶囊(批号:16010157)中利福布汀含量的RSD为0.92%(n=6),表明方法重复性好。
2.8 稳定性试验
精密称取利福布汀原料药(批号:17020156)和利福布汀胶囊(批号:16010157)内容物适量,分别按“2.2.1”项下方法制成供试品溶液,放置于室温,分别在0、2、4、8、10、12 h进样测定,计算各杂质的峰面积。结果,原料药中利福布汀峰面积的RSD为0.07%(n=6),胶囊中利福布汀峰面积的RSD为0.10%(n=6),表明原料药和胶囊的供试品溶液在12 h内稳定。
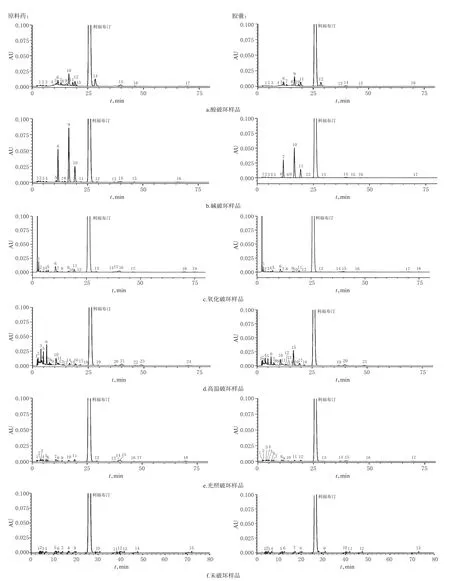
图2 专属性试验的高效液相色谱图Fig 2 HPLC chromatograms of specification test
2.9 耐用性试验
微调流动相流速(0.9、1.1 mL/min)、流动相中乙腈比例(45%、55%)、流动相 pH(6.3、6.7)、柱温(25、35 ℃)、不同色谱柱(Kromasil C8、Sepax C8,规格:均为250 mm×4.6 mm,5 μm),用系统适用性溶液按上述条件进行耐用性试验。结果,各峰的分离度良好,表明方法耐用性较好。耐用性试验结果见表1。

表1 耐用性试验结果Tab 1 Results of durability tests
2.10 有关物质检查法
精密量取“2.2”项下3种溶液各20 μL,分别注入液相色谱仪,记录色谱图至主成分峰保留时间的3倍。供试品溶液色谱图中如有杂质峰,单个杂质峰面积不得大于对照溶液主峰面积(1.0%),大于对照溶液主峰面积0.5倍(0.5%)的峰不得多于1个。各杂质峰面积的和不得大于对照溶液主峰面积的3倍(3.0%)。供试品溶液色谱图中小于灵敏度溶液主峰面积的峰可忽略不计(0.05%)。
2.11 与现行方法比较
2.11.1 色谱条件 在现行方法的供试品溶液制备中,采用先加乙腈溶解再用流动相稀释的方法,制备的质量浓度约为0.5 mg/mL;而新方法的供试品溶液制备时,直接用流动相溶解并稀释制成质量浓度约为1.0 mg/mL的溶液。此新方法中的供试品溶液制备方法,可适当消除溶剂效应,且在进样体积相同的情况下将样品量提高了1倍,这将更有利于杂质的检出。另外,在现行方法中,采用C18柱进行分离,而改进方法参照USP、EP的方法,选用C8柱进行试验。
2.11.2 系统适用性试验 取利福布汀原料药(批号:17020156)按“2.1.2”项下方法制备系统适用性试验溶液,采用现行方法和新方法,进样分析,结果见图1A、B。
由图1可见,在现行方法中,利福布汀峰与相邻的降解峰3之间的分离度为3.47,远小于新方法中利福布汀峰与降解峰3的分离度7.50;另外,采用现行方法时,利福布汀主峰的保留时间约为30 min,较新方法的24 min更长;故新方法的色谱分离能力优于现行方法,且检测时间更短。
2.11.3 样品中有关物质的检查 按“2.10”项下方法测定原料药和胶囊的有关物质(峰面积归一化法),采用现行方法和改进方法进行检查的结果见表2、表3,色谱图见图3、图4。

表2 采用现行方法检查利福布汀及胶囊中有关物质的结果Tab 2 Determination of related substances in rifabutin crude drug and capsules by current methods

表3 采用新方法检查利福布汀及胶囊中有关物质的结果Tab 3 Results of related substances in rifabutin crude drug and capsules by new methods
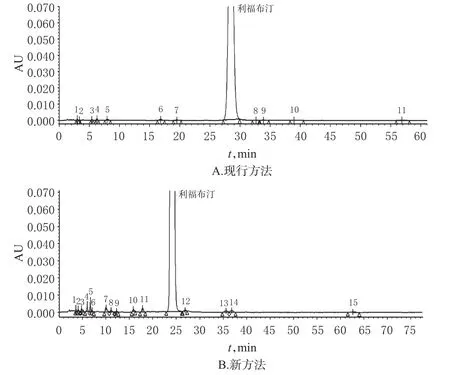
图3 原料药中有关物质检查高效液相色谱图(批号:15070356)Fig 3 HPLC chromatograms of related substance in crude drug(No.15070356)
由表2、表3及图3、图4可见,采用新方法检出的杂质个数较现行方法多1~5个,杂质总量高出0.19%~0.55%,新方法与现行方法比较主峰峰形更好,与杂质峰分离更好,检出的杂质个数更多。

图4 胶囊中有关物质检查高效液相色谱图(批号:17090257)Fig 4 HPLC chromatograms of related substance in capsules(No.17090257)
3 讨论
3.1 色谱条件的选择
3.1.1 色谱柱的选择 现行方法采用C18柱进行分离,而新方法参照USP和EP的方法,选用C8柱进行试验,且色谱柱规格选用了国内常见的250 mm×4.6 mm。由系统适用性试验结果可见,在利福布汀主峰与其前相邻杂质峰的分离方面,新方法的C8柱优于现行方法的C18柱。
3.1.2 溶剂的选择 在现行方法中,基于利福布汀在乙腈中易溶的特点,所以选用乙腈为溶剂溶解样品后再用流动相稀释至刻度的方法。在新方法中,虽然制备的供试品溶液质量浓度更高,但只用流动相依然能够很好地溶解样品。故最终只选用流动相作为溶剂,消除了先加入乙腈后带来的溶剂效应。
3.2 对样品破坏试验的探讨
从强制破坏试验结果可以看出,利福布汀原料药在试验设计的光照、氧化、酸、热和碱降解条件下均发生降解。在按峰面积归一化法计算杂质含量时,笔者发现利福布汀在氧化降解条件下相对保留时间约为0.41的杂质比未破坏时约增加了0.5%;在酸降解条件下相对保留时间约为0.64的杂质比未破坏时约增加了1.2%;在高温降解条件下相对保留时间约为0.19、0.25和0.41的杂质比未破坏时分别增加了0.5%、1.1%和0.5%;在碱降解条件下相对保留时间约为0.45、0.64和0.75的杂质比未破坏时分别增加了2.4%、4.7%和1.5%。故认为对试验中主要降解产物的结构分析有待进一步研究,以便更好地控制利福布汀的质量。
综上,采用新方法进行利福布汀原料药和胶囊中有关物质的检查,方法适用性良好,且与现行方法相比较,有出峰更快、峰形更好、分离杂质更多的优点。故新的方法灵敏、简单,适用于对利福布汀原料药及胶囊的有关物质检查。
