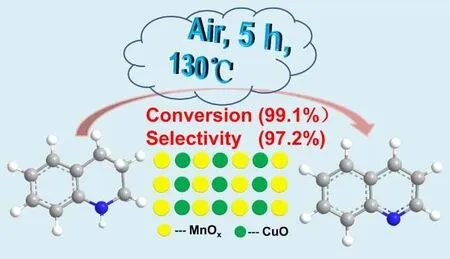
Cu2-MnOx高效催化1,2,3,4-四氢喹啉氧化脱氢芳构化
2019-07-26陈福山赵松林杨涛江涛涛倪珺张群峰李小年
陈福山,赵松林,杨涛,江涛涛,倪珺,张群峰,*,李小年,*
1浙江工业大学工业催化研究所,杭州 3100142九江学院江西省生态化工工程技术研究中心,江西 九江 3320053台州学院医药化工学院,浙江 台州 318000
1 引言
含氮杂环芳族化合物是常见的药物和生物相关的中间体分子1,2。含氮杂芳烃制备的典型方法是从饱和杂环化合物氧化脱氢。在氧化脱氢过程中催化剂是不可缺少重要因素之一,可分成均相催化剂和多相催化剂。到目前为止,均相催化剂主要有Ir3,4、Ru5、Fe6及B7等的络合物。然而,均相系统使得分离催化剂和净化产物变得复杂,难以实现高催化性效率。而且催化剂难以回收再利用,可持续性和成本效益受到很大的挑战。在这种情况下,非均相催化剂非常有吸引力,它们具有可重用性,低成本和工艺简单等优点。一些Rh8、Ru9、Pd10、Pt11、Au12、Fe13、Cu14基等非均相催化剂在N-杂环的氧化脱氢中也进行了相关的研究。但是,这些催化体系绝大部分需要贵金属、添加剂或苛刻的反应条件及催化剂制备复杂等。
最近有文献报道,价格低廉的锰基多相催化剂在THQL氧化脱氢芳构化反应中具有优异的催化性能,而且不要其它添加剂,仅以空气或氧气为氧化剂15,16。但这种催化剂制备的方法较为复杂,催化性能也需进一步提高。
采用新型无模板草酸盐合成方法,可以合成高表面积、催化活性较高的介孔锰氧化物催化剂,但主要应用于环境方面深度氧化17-20。而在有机化合物的选择性催化氧化鲜有报道。本文采用草酸铵共沉淀的方法制备了CuO掺杂的无定型MnOx催化剂,并探究了CuO的掺杂对THQL氧化脱氢芳构化催化性能的影响。
2 实验部分
2.1 催化剂制备
参照文献19,20,用草酸铵共沉淀路线合成催化剂。
称取0.04 mol (14.316 g) Mn(NO3)2(50% (w,质量分数))溶液(阿拉丁公司,AR),0.004 mol M(NO3)2-3·xH2O (M = Fe、Ni、Co、Al、Cu,x代表结晶水的分子数) (Fe(NO3)3·9H2O,98.5%、Ni(NO3)2·6H2O,98.0%、Co(NO3)3·6H2O,99.0%、Al(NO3)3·9H2O,99.0%、Cu(NO3)2·3H2O,99.0%,都为AR且购自于国药公司)溶于190 mL高纯水(16 MΩ),再称取0.0528 mol (7.503 g)的(NH4)2C2O4[nMn+ nM) :n((NH4)2C2O4) = 1 : 1.2] (阿拉丁公司,99.8%,AR)溶解于200 mL高纯水。在室温下强烈搅拌下(700 r·min-1)将(NH4)2C2O4澄清溶液快速加入到锰和其它金属离子的混合溶液中。在反应期间,草酸盐沉淀物迅速形成,搅拌40 min后,将沉淀物(Precursor)过滤,随后用高纯水和无水乙醇(天津大茂,AR)洗涤数次,然后在80 °C下干燥20 h。将样品转移到坩埚中,在马弗炉(杭州蓝天,SXQF-4-10)中以2 °C·min-1加热至一定温度350 °C,然后再处理5 h,制得样品用M1-MnOx表示(M = Fe、Ni、Co、Al、Cu)。在其它条件不变的情况下,Cu(NO3)2·3H2O为0、0.008、0.012、0.016 mol,相应(NH4)2C2O4的量符合[n(Mn + M) : n((NH4)2C2O4) = 1 : 1.2,对应的催化剂分别用MnOx、Cu2-MnOx、Cu3-MnOx、Cu4-MnOx表示。Cu(NO3)2·3H2O为0、0.008 mol,对应的水合草酸锰前驱体分别用Precursor-1和Precursor-2表示。Precursor-2在其它焙烧条件不变的情况下,在450和550 °C焙烧的样品分别用Cu2-450和Cu2-550表示。
2.2 催化剂表征
利用PNAlytical公司X′Pert Pro型X射线衍射仪(Ni滤波,Cu Kα辐射源,工作电压40 kV,工作电流30 mA)进行样品晶体结构测定,2θ = 0°-10° (小角),2θ = 10°-80° (广角)。利用Micromeritics公司的ASAP-2020型表面性质分析仪对样品进行织构性质测定,吸附测定前,样品先在200 °C脱气预处理4 h,样品的比表面积是通过Brunauer-Emmett-Teller (BET)方法求得21。利用扫描电子显微镜(SEM)观察样品的表观形貌(TESCAN VEGA II LSU)。利用透射电镜(TEM)查看样品的细微结构(Philips-FEI公司,Tecnai G2 F30S-Twin,300 kV),利用其配套的能谱仪(EDX)对样品元素进行定性分析。利用实验室自制氢气还原装置(TCD检测器)(H2-TPR),用流速30 mL·min-1的5%-H2/Ar混合气,以10 °C·min-1从30至600 °C对0.1 g样品氢气还原能力进行测定。利用Kratos AXIS ULtra DLD X射线光电子能谱(XPS)用于分析样品的表面组成和化学状态。在分析之前,将样品在超高真空脱气,然后通过使用单色Al KαX射线源进行XPS实验,结合能参考在284.8 eV的C 1s峰。利用原子吸收光谱仪(AAS)对样品Cu含量进行测定(Perkin-Elmer,PinAAcle 900T)。
2.3 催化剂活性评价
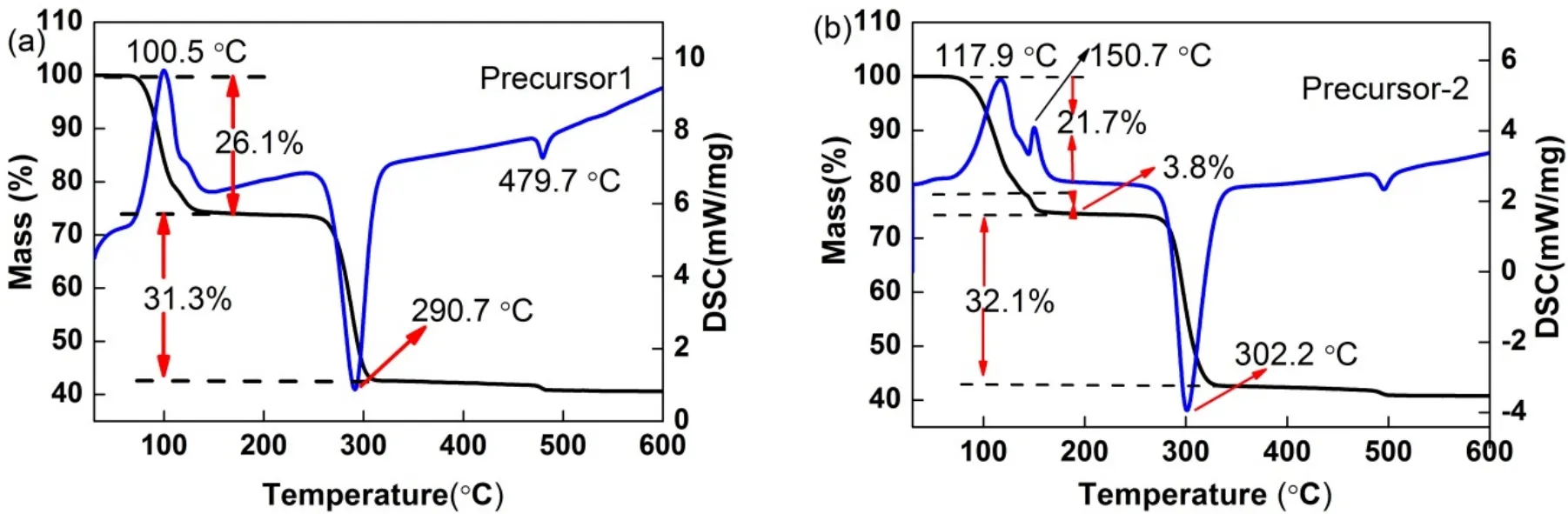
图1 (a) Precursor-1和(b) Precursor-2热分解的TG及DSC分析Fig.1 TG and DSC (thermogravimetry and heat flow) analyses for the decomposition of(a) Precursor-1 and (b) Precursor-2.
在配有冷凝装置的25 mL单颈圆底烧瓶中加入0.25 mmol (31.4 μL) 1,2,3,4-四氢喹啉、5 mL二甲基甲酰胺(DMF)、25 mg催化剂,使用气球的高纯空气作为氧化剂。油浴温度130 °C,磁力搅拌转速为700 r·min-1,反应5 h。待反应结束后,冷却混合物过滤除去催化剂。通过GC-MS (Thermo Fisher TRACE1310-TSQ8000Evo)和标准物质鉴定产物混合物。通过GC(Agilent 6890N,FID)使用面积归一化方法确定转化率和选择性。
3 结果和讨论
3.1 TG-DSC
图1a,b显示Precursor-1和Precursor-2的TGDSC曲线。图1a在100.5 °C时的第一次重量损失对应于前体脱水为无水草酸锰,这表明Precursor-1主要是含水的草酸锰17,20。Precursor-1在100.5 °C时的重量损失为26%。在290.7 °C的第二次重量损失归属于草酸锰分解成锰氧化物,在该步骤中重量损失为31%19,22。479.7 °C第三次重量损失有一个放热峰,这意味着有一个相变,这暗示该前驱体是MnC2O4·3H2O23。Precursor-2的TG-DSC曲线表明,掺Cu以后Precursor-2分解和相变温度都向高温漂移,说明Cu的掺杂影响了水合草酸锰的分解(图1b)。由于Cu实际掺入量非常少(0.81% (质量分数,w)),因此水合草酸铜的TG和DSC峰被水合草酸锰覆盖,不能有效显示出来。在150.7 °C新出现一个明显的吸热峰可能是受到掺杂CuO的影响,小部分水合草酸锰脱水温度向高温偏移所致,重量损失仅为3.8%。
3.2 XRD
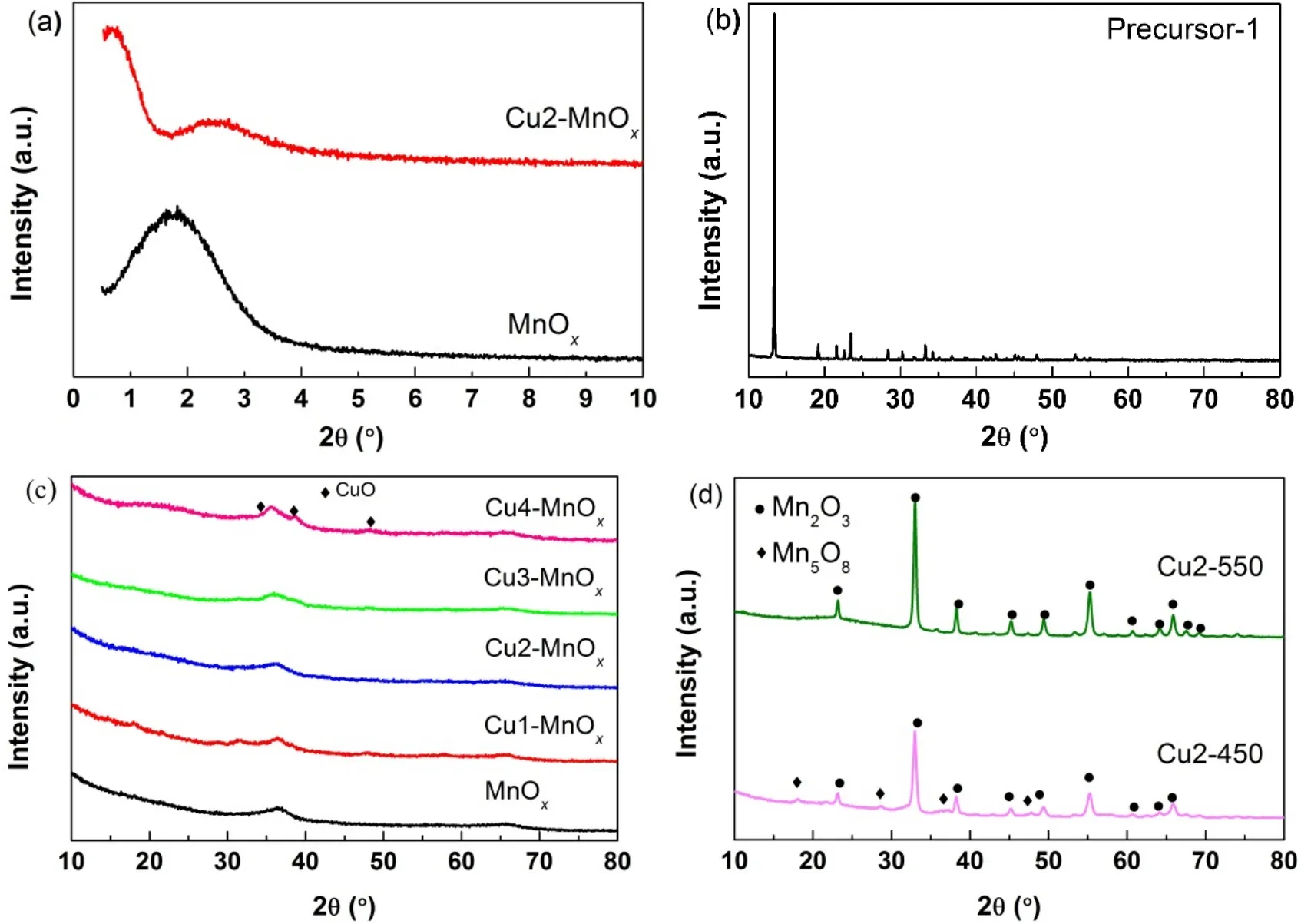
图 2 (a)样品MnOx和Cu2-MnOx的小角衍射(0.5°-10°) XRD图谱,(b-d) Precursor-1和样品的广角衍射(10°-80°) XRD图谱Fig.2 (a) XRD patterns of the MnOx and Cu2-MnOx samples at low angle (0.5°-10°),(b-d) wide angle XRD patterns of Precursor-1 and the samples.
图2 a显示样品MnOx和Cu2-MnOx小角衍射XRD图谱。MnOx在0.5°-4°显示一个大宽峰,在掺Cu以后Cu2-MnOx出现两个峰,其中在2°左右峰变弱,另一个峰向低角度发生偏移的现象,说明这种材料在掺少量Cu以后孔结构发生变化24。图2b显示Precursor-1的广角XRD图谱,归属于MnC2O4·3H2O (PDF# 32-0648),再次确认了TGDSC的结果。图2c显示MnOx、Cu1-MnOx、Cu2-MnOx、Cu3-MnOx及Cu4-MnOx的广角XRD图谱。MnOx显示为无定型的结构,这和文献报道基本一致19,20。在掺杂了少量CuO后,Cu1-MnOx、Cu2-MnOx、Cu3-MnOx主要保持无定型的结构,同时出现很弱的亚稳态Mn5O8相(PDF# 39-1218)的衍射峰,说明少量CuO的掺杂并没有改变锰氧化物主要的无定型结构,且没有明显的CuO的衍射峰出现,暗示CuO高度分散。继续增加CuO的掺杂量,Cu4-MnOx中锰氧化物继续保持主要的无定型的结构,但出现了部分较弱的衍射峰,这和CuO的衍射峰基本一致(PDF# 44-0706)。图2d显示在450和550 °C焙烧Precursor-2所获样品的广角XRD图谱。随着焙烧温度的升高,样品由无定型的结构变成晶体结构(图2c (Cu2-MnOx),图2d)。图2d (Cu-450)的衍射峰归属于Mn2O3(PDF# 41-1442)和少量的Mn5O8(PDF#39-1218)晶相,图2d (Cu-550)仅显示增强了的Mn2O3衍射峰,说明随着焙烧温度进一步升高,样品的晶粒粒径进一步增大,而且都没有出现明显CuO的衍射峰。
3.3 N2吸附脱附
图3a显示样品的N2物理吸附-脱附等温线。N2吸附和脱附等温线显示七种样品均为IV型等温线(IUPAC)21,其中Cu2-MnOx为典型的H3型回滞环,MnOx、Cu1-MnOx、Cu3-MnOx和Cu4-MnOx介于H3型和H4型之间,Cu2-450、Cu2-550为典型的H1型回滞环,说明Cu2-MnOx的介孔结构比MnOx、Cu1-MnOx、Cu3-MnOx和Cu4-MnOx更加明显,Cu2-450、Cu2-550为典型的介孔结构21,25。MnOx、Cu1-MnOx、Cu2-MnOx、Cu3-MnOx和Cu4-MnOx五种样品的等温线中,在P/P0( < 0.02)的N2的急剧吸附是由于少量微孔的N2填充,表明这四种样品均有部分微孔存在20。
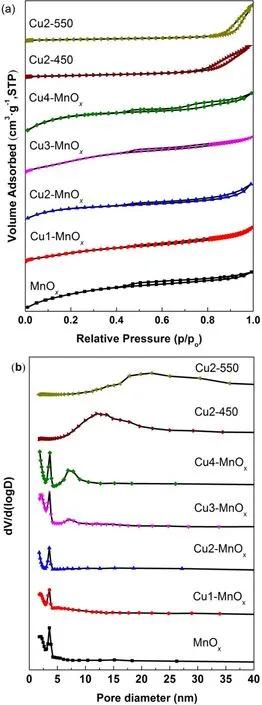
图3 (a)样品氮气吸附-脱附等温线,(b)样品BJH脱附支孔径分布Fig. 3 (a) Nitrogen adsorption-desorption isotherms of the samples, (b) BJH (Barrett-Joyner-Halenda)desorption pore size
图3 b和表1显示五种样品的孔径分布和织构性质。从图3b的孔径分布可以看出MnOx、Cu1-MnOx、Cu2-MnOx、Cu3-MnOx和Cu4-MnOx五种样品均具有介孔和微孔结构,Cu2-450、Cu2-550为明显的介孔结构,这与N2吸附和脱附等温线分析结果基本一致。表1孔径由BJH方法从N2脱附分支中获得,孔容由BJH方法从N2脱附分支累积的孔隙体积获得。从表1看出,MnOx的BET比表面积是289.1 m2·g-1、孔容0.17 cm3·g-1、最小的平均孔径3.2 nm。少量CuO掺入使锰氧化物的孔径增加,其中Cu2-MnOx平均孔径增加至最高4.9 nm,BET比表面积(239.3 m2·g-1)和孔容(0.16 cm3·g-1)略有降低。较大的孔径促进了反应物分子吸附和产物分子的脱附。增加CuO的掺杂量,表1显示Cu3-MnOx平均孔径降低至3.4 nm,BET比表面积增加至最大(302.9 m2·g-1),孔容(0.22 cm3·g-1)也增加至最大,这可能是由于CuO的掺杂量增加导致锰氧化物的微棒结构出现破裂且出现部分小颗粒所致,这也得到SEM确认(图4e)。继续增加CuO的掺杂量,表1(Cu4-MnOx)显示平均孔径又增加至4.0 nm,BET比表面积(246.2 m2·g-1)又明显降低,孔容(0.13 cm3·g-1)也明显降低,这可能是由于大量CuO的掺杂导致锰氧化物的微棒结构出现较大幅度的破碎且出现大量不规则的大颗粒物所致,这也得到SEM证实(图4f)。Cu3-MnOx、Cu4-MnOx在6.9 nm附近出现少量介孔结构(图3b,Cu3-MnOx,Cu4-MnOx),这可能是由于CuO的过量掺杂导致锰氧化物的部分孔道发生坍塌形成较大的孔所致。高温焙烧的Cu2-450和Cu2-550比表面积分别大幅降低至47.和27.1 m2·g-1,平均孔径分别增加至14.7和22.7 nm,孔容分别是0.16和0.17 cm3·g-1。
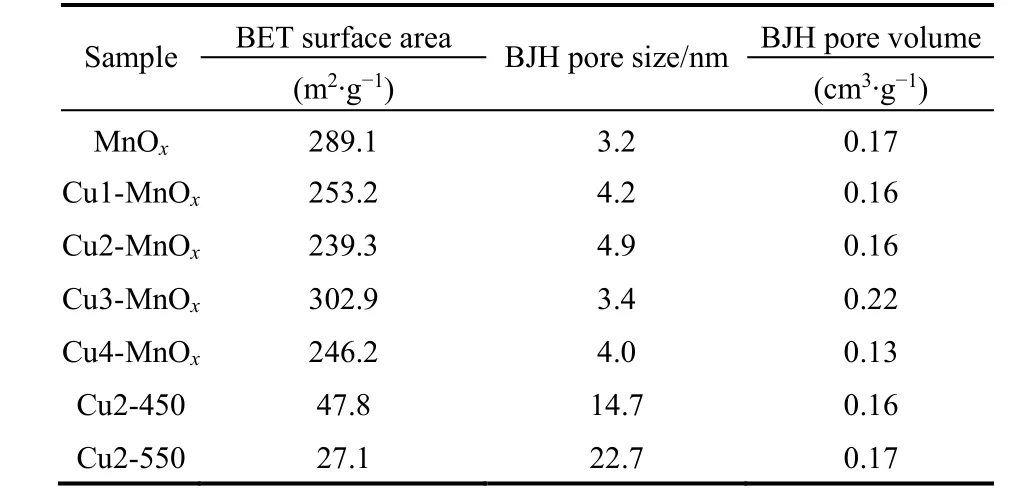
表1 样品的织构性质Table 1 Textural properties of the samples.
3.4 SEM
图4显示Precursor-1、MnOx、Cu1-MnOx、Cu2-MnOx、Cu3-MnOx,、Cu4-MnOx的SEM图。从图4a,b可以看出,在焙烧后Precursor-1在失重57.4%后(图1a),基本保持了前驱体微棒状形貌,说明焙烧后的材料具有较大的孔隙率。图4b显示几乎全是微棒,图4c-d显示基本和图4b类似,表明少量掺CuO对锰氧化物的形貌基本没有改变。在提高CuO的掺杂量后,图4e基本还是保持图4b的微棒状,但同时出现了少量小颗粒。图4f显示微棒数量减少和部分发生破裂,有大量的不规则颗粒生成。通过对颗粒物EDS分析,主要是锰氧化物,而不是氧化铜,这说明氧化铜不是新出现独立的颗粒物,而是和锰氧化物共同存在。
3.5 TEM
图5a和5c的HRTEM图说明MnOx和Cu2-MnOx是无定型的结构。图5b和5d的SAED较暗的环状图案,确认了MnOx和Cu2-MnOx无定型的结构,这个结果也和前面XRD的结果一致。Cu2-MnOx的STEM(图5e)和EDX(图5i)说明样品不仅存在Mn和O两种元素,而且有Cu元素的存在。而且没有看到CuO的颗粒存在,说明Cu元素高度分散,这个结果也得到O、Mn、Cu元素mapping的进一步确认(图5f-h),也和Cu2-MnOx的XRD结果一致,没有CuO的衍射峰出现。

图4 (a) Precursor-1, (b) MnOx, (c) Cu1-MnOx, (d) Cu2-MnOx, (e) Cu3-MnOx, (f) Cu4-MnOx的SEM图Fig. 4 SEM images of (a) Precursor-1, (b) MnOx, (c) Cu1-MnOx, (d) Cu2-MnOx, (e) Cu3-MnOx, (f) Cu4-MnOx.
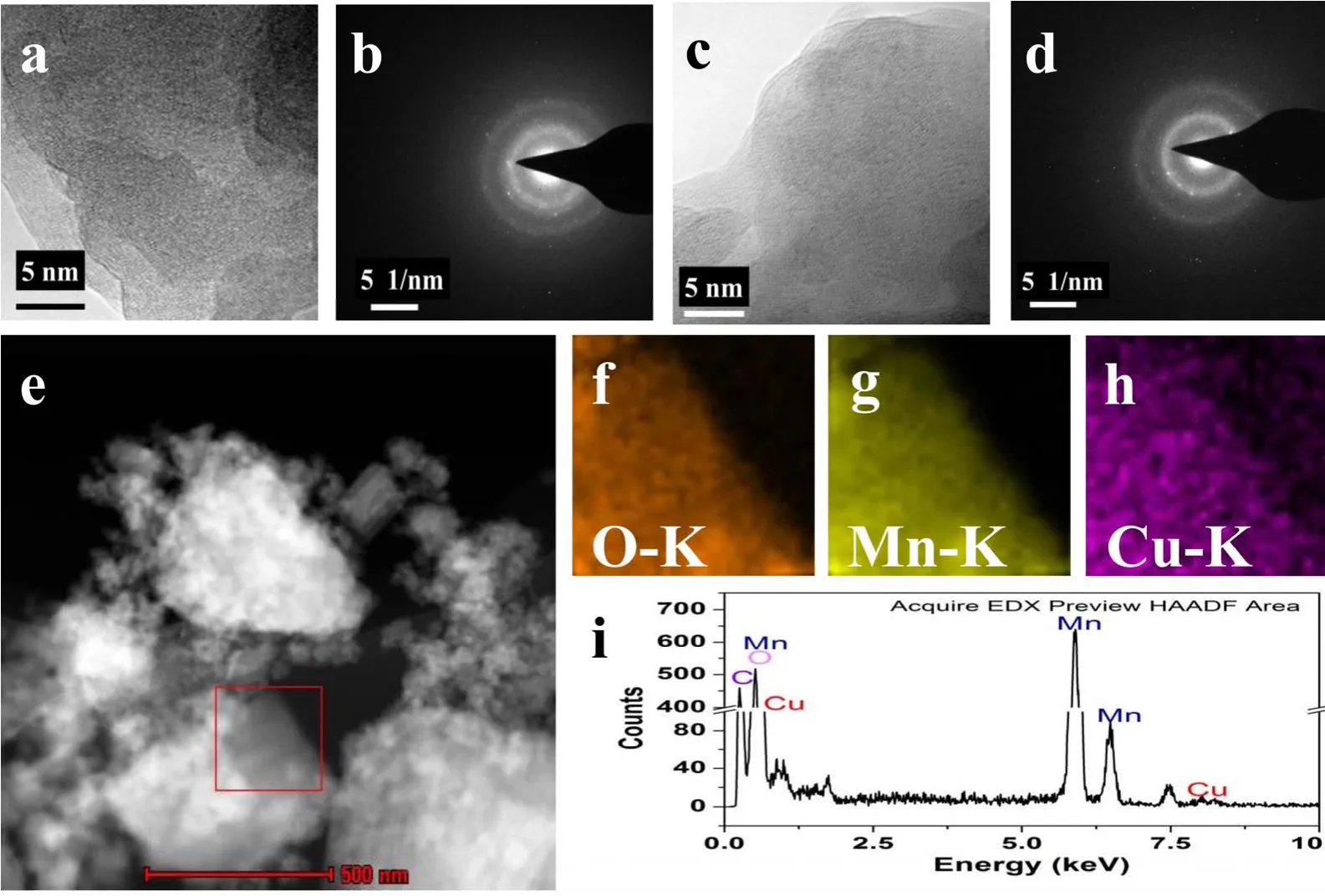
图5 (a,c) MnOx和Cu2-MnOx的HRTEM图,(b,d) MnOx和Cu2-MnOx的SAED图,(e) Cu2-MnOx的STEM图,(f-h) Cu2-MnOx相应元素的mapping图,(i) Cu2-MnOx的EDX分析图Fig. 5 (a, c) HRTEM images of MnOx and Cu2-MnOx, (b, d) SAED of MnOx and Cu2-MnOx, (e) STEM image of Cu2-MnOx, (f-h) corresponding elemental mapping images of the Cu2-MnOx, and (i) EDX analysis of Cu2-MnOx.HRTEM: high resolution transmission electron microscope; SAED: selected area electron diffraction; STEM: scanning transmission electron microscopy;
3.6 XPS
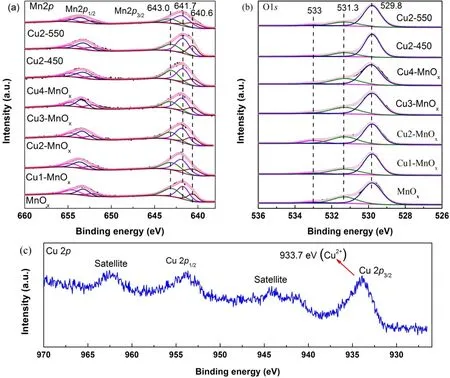
图6 (a) 样品Mn 2p的XPS谱图,(b)样品O 1s的XPS谱图,(c) Cu4-MnOx的Cu 2p XPS谱图Fig. 6 (a) Mn 2p XPS spectra of the samples, (b) O 1s XPS spectra of the samples, (c) Cu 2p XPS spectra of the Cu4-MnOx.
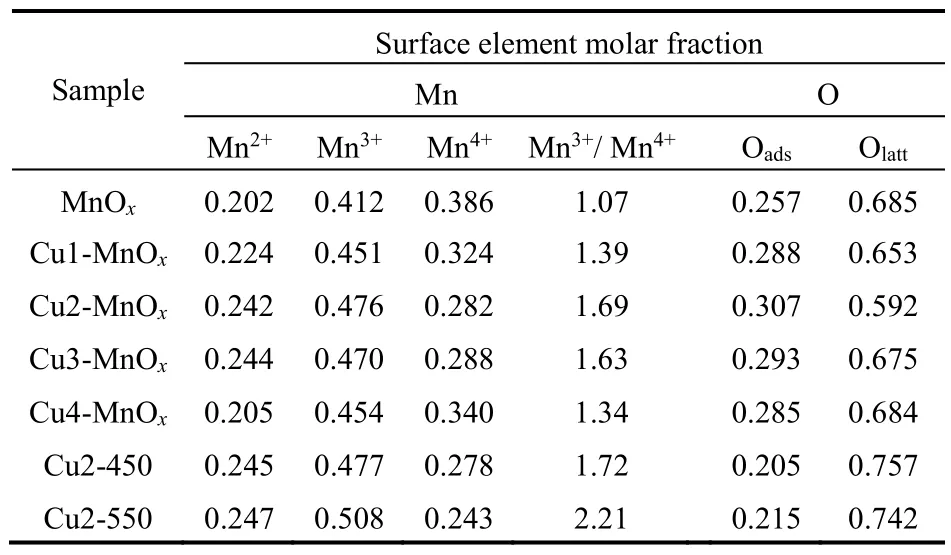
表2 从Mn 2p和O 1s XPS光谱得出的表面不同元素组成的摩尔分数Table 2 Summary of molar fraction of different elemental components on surface from Mn 2p and O 1s XPS spectra.
为了进一步研究样品的价态信息及组成,图6a-c显示了样品的Mn 2p、O 1s及Cu 2p XPS光谱。Mn 2p XPS光谱在640.6、641.7和643.0 eV处有三个特征峰,分别归属于催化剂表面的Mn2+,Mn3+和Mn4+离子(图6a)19,26。在Mn 2p3/2和Mn 2p1/2状态之间显示出11.5 eV的能量分离。从表2样品的Mn 2p XPS光谱表面元素摩尔分数可以看出,掺杂CuO的无定型结构锰氧化物的Mn3+的比例明显提高,其中Cu2-MnOx和Cu3-MnOx的表面Mn3+含量分别高达0.476和0.470。表面Mn3+的比例在THQL催化氧化脱氢中是重要的活性位,催化剂Mn3+比例越高,催化活性也越高15,16。与此同时Cu2-MnOx(表2,Cu2-MnOx)具有最高的Mn3+/Mn4+(1.69)。从图6b O 1s XPS光谱可以看出,O 1s光谱非常宽且不对称,这可能是由于各种氧物种的叠加形成的。O 1s XPS光谱可以在529.8、531.3、533.0 eV处拟合成三个峰,分别归属于晶格氧(O2-,表示为Olatt),表面吸附氧(O-2,O22-,O-,表示为Oads),表面氧(吸附的OH基团,分子水和碳酸盐物质) (表示为Osuf)15,16。从表2样品的O 1s XPS光谱表面元素摩尔分数看出,所有样品的晶格氧含量远高于其它氧物种的含量。吸附氧(Oads)在催化氧化过程中是非常重要的27-29,我们希望通过CuO的掺杂使无定型的锰氧化物具有较多的吸附氧。表2显示无定型结构的Cu2-MnOx(Oads含量0.307)和Cu3-MnOx(Oads含量0.293)具有较多的吸附氧物种,这意味着这种催化剂在有机选择性氧化反应中具有较好的活性氧的流动性和可利用性27,30,31。高温焙烧的有较好结晶度和低比表面积的Cu2-450 (Mn3+含量0.477)和Cu2-550 (Mn3+含量0.508)的表面Mn3+含量进一步提高,但Cu2-450 (Oads含量0.205)和Cu2-550 (Oads含量0.215)具有较低的吸附氧含量,这主要是较高结晶度的锰氧化物结构缺陷减少导致吸附氧的数量大幅降低30,暗示它们在有氧氧化反应中可能具有较弱的催化活性。通过比较我们发现,无定型锰氧化物Mn3+含量高的样品一般都有较高含量的吸附氧,暗示在无定型锰氧化物中吸附氧的产生可能主要与Mn3+有关。图6c显示了Cu4-MnOx的Cu 2p XPS光谱,Cu 2p3/2的结合能在933.7 eV和卫星峰的存在说明Cu元素是以+2价存在,这也和XRD测试结果完全一致32。
3.7 H2-TPR
图7显示了样品在300至600 °C氢气程序升温还原图。基于Kapteijn等提出的锰氧化物的H2还原理论19,33,34,锰氧化物的连续还原过程如下:MnO2(Mn2O3) → Mn3O4→ MnO,MnO在30至600 °C是最终的还原态。MnOx、Cu1-MnOx、Cu2-MnOx、Cu3-MnOx样品在281、271、259、198 °C的还原峰归属于MnO2→ Mn4O3的还原,在296、288、276、229 °C的还原峰归属于Mn2O3→ Mn3O4的还原,在452、435、409、261 °C的还原峰归属于Mn3O4→MnO的还原。可以看出随着CuO掺杂量的增加各种锰氧化物的还原温度进一步向低温偏移。从图2c (Cu1-MnOx、Cu2-MnOx、Cu3-MnOx) XRD可以看出没有出现CuO的衍射峰,说明在这些样品中CuO是高分散的。高分散CuO的还原峰温度是214 °C35,由于实际掺杂进入锰氧化物中的CuO含量较低(其中Cu3-MnOx的Cu含量2.83%(w)),因此高分散CuO的较弱的还原峰可能被MnO2→Mn4O3还原峰所覆盖。在Cu4-MnOx中由于CuO的掺杂量大幅增加,由图2c (Cu4-MnOx) XRD可以看出,出现了明显CuO的衍射峰,说明CuO已经由高分散转变成晶体颗粒,从而导致Cu4-MnOx的还原温度和Cu3-MnOx相比向高温偏移34,35,在264 °C大宽峰是不同价态锰氧化物和CuO还原峰重叠形成的。和低温焙烧的样品相比,高温焙烧的Cu2-450和Cu2-550分别在312和355 °C出现一个各种锰氧化物和CuO还原的宽峰,而且还原温度和低温焙烧的样品相比,明显向高温偏移,这是因为较高结晶度的锰氧化物具有较高的还原温度19。锰氧化物的还原温度降低主要是CuO的掺入形成了CuO和MnOx的协同作用及CuO和锰氧化物界面产生的氢溢流所致34,35。文献35报道掺CuO的锰氧化物用CO还原,还原温度也相似的向低温偏移,说明少量CuO的掺入确实降低了锰氧化物的还原温度。锰氧化物的还原温度和晶格氧的流动性直接相关,还原温度越低,晶格氧的流动性越高,有氧反应催化活性也越高36。Cu3-MnOx具有最低还原温度,暗示其拥有最高的晶格氧流动性和较高的有氧反应催化活性。
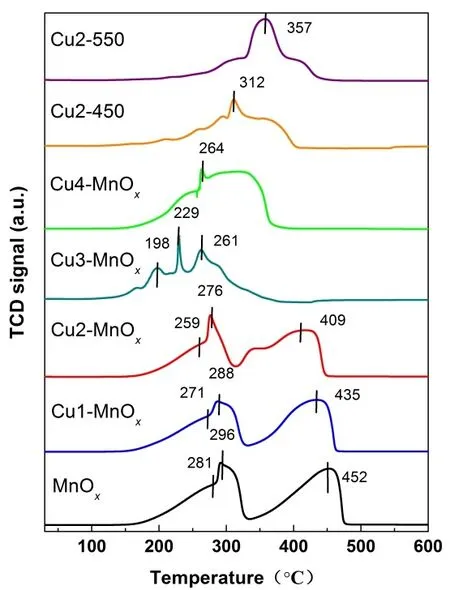
图7 样品30至600 °C的氢气程序升温还原图谱Fig. 7 H2-TPR profiles of the samples from 30 to 600 °C.
3.8 催化剂的催化性能
为了制备高效的THQL芳构化催化剂,通过对MnOx.进行不同金属氧化物的掺杂来改善其催化性能。表3显示掺杂不同金属氧化物的锰氧化物催化剂对催化THQL芳构化活性比较。在没有催化剂的参与下,THQL只有极少量的转化(0.9%) (表3 Entry1)。MnOx在不掺杂金属氧化物的情况下,THQL转化率为90.8%,QL的选择性85.2%,唯一的副产物是喹啉氮氧化物(表3 Entry2)。在掺杂其它金属氧化物后,Cu1-MnOx的转化率(97.1%)和选择性(92.7%)都显著提高(表3 Entry7)。而Ni1-MnOx催化性能明显下降,具有最低的转化率(85.1%)和选择性(82.4%) (表3 Entry5)。
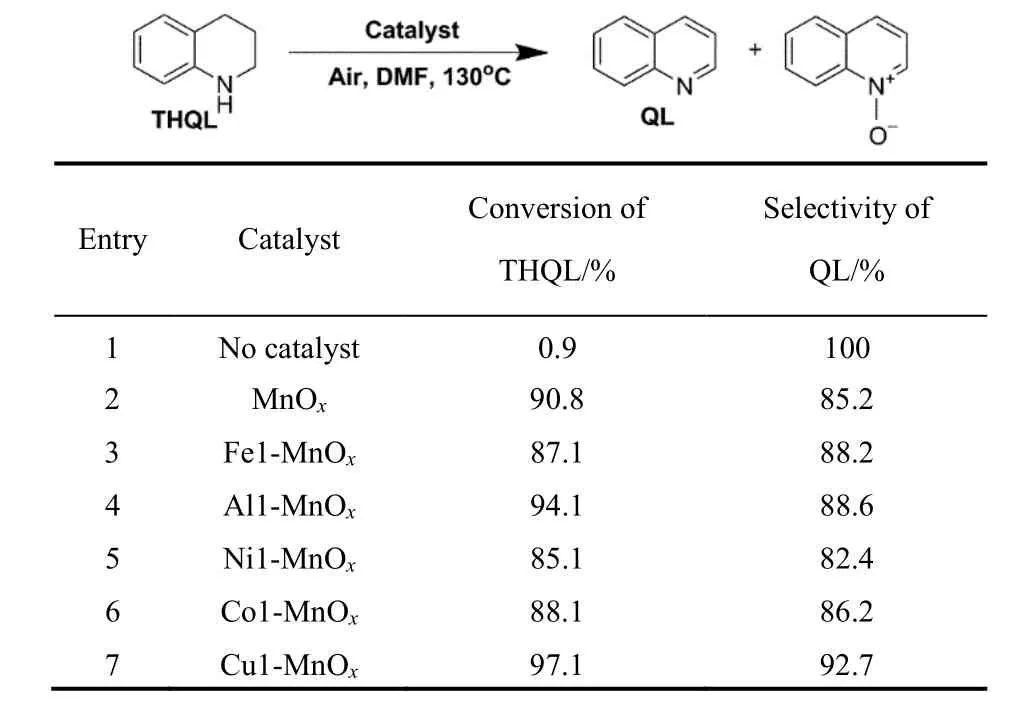
表3 掺杂不同金属氧化物锰氧化物催化剂对THQL芳构化催化活性比较Table 3 Comparison of catalytic activity of THQL aromatization by MnOx catalysts with doped different metal oxide.
在确定了Cu作为最佳的掺杂元素后,不同Cu的掺杂量被进行了筛选。表4显示了掺杂不同CuO量的MnOx催化剂对THQL芳构化催化活性比较。当Cu的掺杂量是0.81% (w)时,Cu2-MnOx具有最高的转化率(99.1%)和选择性(97.2%) (表4 Entry2)。继续增加Cu的掺杂量,Cu3-MnOx(2.83%(w) Cu)也具有较高的转化率(98.1%)和选择性(96.9%) (表4 Entry3),而Cu4-MnOx(8.92%(w) Cu)的转化率(97.3%)略微降低和选择性(93.0%)明显降低(表4 Entry4)。而由草酸铵和硝酸铜在相同条件下制备的CuO具有非常低的催化活性,转化率和选择性分别是10.6%、88.3% (表4 Entry5)。由表3 (Entry2)和表4 (Entry1-5)说明在CuO掺杂的锰氧化物催化剂中CuO和MnOx产生了较强的协同作用,促进了THQL的芳构化,这也在各种表征结果中得到了确认。当Precursor-2在450和550 °C焙烧后,Cu2-450、Cu2-550的催化活性大幅度下降,转化率分别为37.5%、30.4% (表4,Entry6,7)。此外从表4可以看出,和其它掺CuO催化剂相比,以空气为氧化剂在DMF溶剂中,Cu2-MnOx具有最高的TOF(turnover frequency)值:0.00198 mmol·mg-1·h-1。通过构效关系的比较,我们有趣的发现无定型锰氧化物中Mn3+和吸附氧的含量越高催化活性也越高,Mn4+的含量可能与副产物喹啉氮氧化物生成有关,Mn3+/Mn4+越高喹啉的选择性也越高,这个结果也进一步得到了文献的确认15。高结晶度的Cu2-450、Cu2-550虽然有很高的Mn3+含量和Mn3+/Mn4+,但由于比表面积和吸附氧含量大幅减低,而且晶格氧的流动性也较低,这可能是其具有较低催化性能的重要原因,这也得到BET、XPS和H2-TPR确认。Cu3-MnOx和Cu2-MnOx相比Mn3+、吸附氧含量及Mn3+/Mn4+都相近,而BET比表面积明显增加,晶格氧流动性明显提高,但转化率和选择性近似,可能与Cu2-MnOx具有比Cu3-MnOx大的平均孔径有关。
在确定了催化剂最佳制备条件后,反应条件也被进一步探索。为了确定最佳氧化剂的类型,其它的气氛被进行测试。在O2气氛下,在反应进行1 h时的转化率(92.9%)明显高于空气气氛1 h的转化率(63.0%),而选择性明显下降至86.4%,这是由于在过量O2下QL过度氧化导致喹啉氮氧化物副产物增加所致,这和文献报道基本一致(表4,Entry8,图8)16。而在N2气氛下,THQL的转化率和QL选择性分别是18.0%、94.6% (表4,Entry9),这主要是Cu2-MnOx的活性晶格氧和吸附氧物种氧化所致,说明O2是THQL芳构化必不可少的氧化剂16,36。由于在氧气气氛中会导致过度氧化产物喹啉氮氧化物的增加,这说明最经济的空气是THQL的氧化脱氢中效果最佳的氧化剂。为了确定最佳的溶剂,DMF、乙腈、甲苯、正己烷和正辛烷分别作为溶剂进行测试。表4 (Entry2,10-13)显示,非极性的甲苯具有最高的转化率(99.9%),但选择性(77.1%)较低,而极性的DMF不仅具有很高的转化率(99.1%),还有最高的选择性(97.2%)。这说明极性的DMF是较为理性的溶剂。转化率和选择性与时间的关系也被进一步考察,图8显示随着反应时间的延长THQL的转化率也逐渐增加,在反应5 h后,转化率达到99.1%,进一步延长反应时间至9 h,转化率基本变化不大,而反应的选择性从反应开始到结束始终变化不大,这说明喹啉氮氧化物量也随QL的增加而增加。
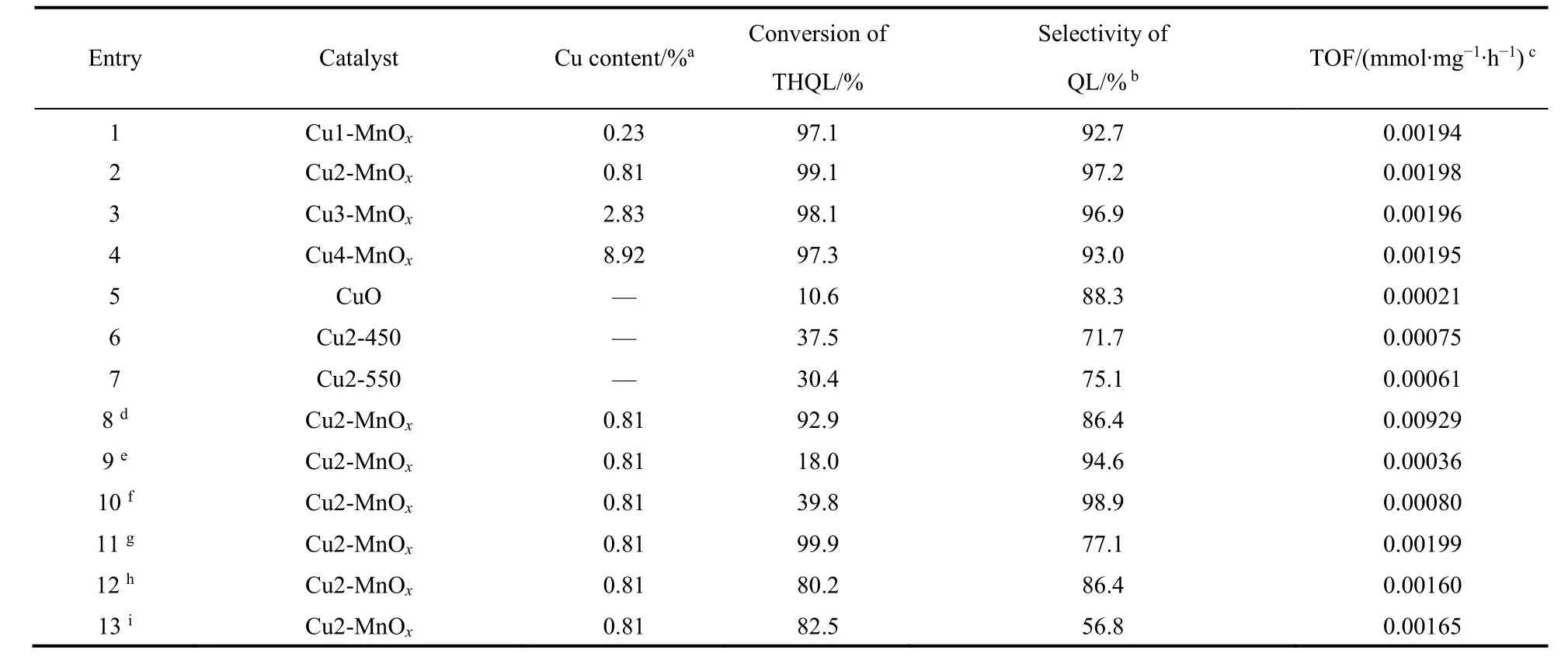
表4 掺杂不同Cu量的MnOx催化剂对THQL芳构化催化活性比较Table 4 Comparison of catalytic activity of THQL aromatization by MnOx catalysts with doping different amount of Cu.
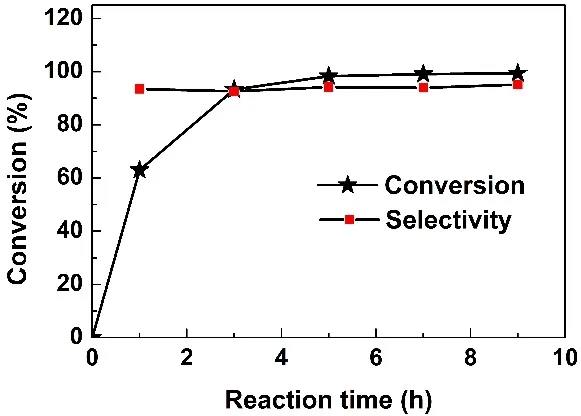
图8 Cu2-MnOx催化活性和时间的关系Fig. 8 Catalytic activity of Cu2-MnOx dependent on the reaction time.Reaction conditions: THQL (0.25 mmol), catalyst (25 mg),DMF (5.0 mL), 130 °C, air balloon.
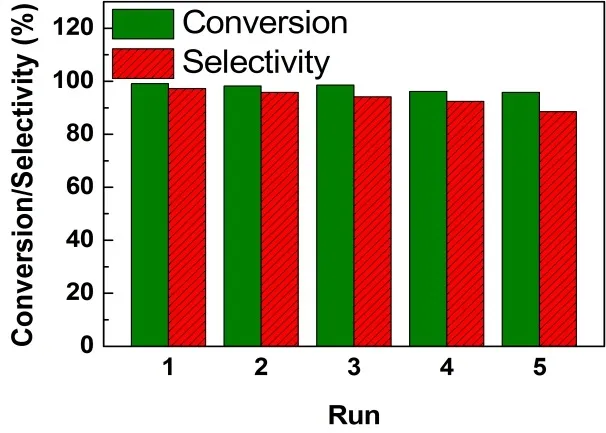
图9 Cu2-MnOx重复性测试Fig. 9 Reusability of the catalyst Cu2-MnOx.Reaction conditions: THQL (0.25 mmol), catalyst (25 mg),DMF (5.0 mL), 130 °C, reaction time (5 h), air balloon.
催化剂的可重复使用性和多相性是多相催化剂的重要性质。反应后,通过离心反应混合物除去反应液,用乙醇洗涤催化剂,然后在250 °C下活化1 h,活化的目的是从催化剂表面除去吸附的物质。图9显示了Cu2-MnOx催化剂的重复性测试结果。从图9可以看出Cu2-MnOx催化剂套用5次后催化活性没有明显降低,催化剂的选择性略有下降,这可能是催化剂随着套用次数的增加,Cu元素的含量有所流失所致,这也得到了第三次套用后催化剂AAS测试确认(Cu2-MnOx中Cu含量0.67% (w))。催化剂的多相性通过热过滤进行测试,从图10可以看出,在转化率为63%时过滤掉催化剂在同样条件下继续反应,反应的转化率并没有进一步的增加,这说明起催化作用的是多相催化剂。
表5总结了文献报道的不同锰基多相催化剂对THQL氧化脱氢芳构化合成喹啉的实验结果。从表5可以看出有关锰基多相催化剂催化THQL的氧化脱氢的已经报道了的能查阅到的文献只有2篇。表5 (Entry1-4)显示AMO、K-OMS-2、Meso-Cs/MnOx、Meso-MnOx的催化活性都较低,但Meso-Cs/MnOx、Meso-MnOx的选择性很高(99%)。Ni2Mn-LDH具有最高的催化活性,TOF值达0.00247 mmol·mg-1·h-1,但喹啉的选择性偏低。而我们课题组制备的Cu2-MnOx不仅具有较高的转化率(99.1%),而且选择性(97.2%)也较高,TOF值也达0.00198 mmol·mg-1·h-1。
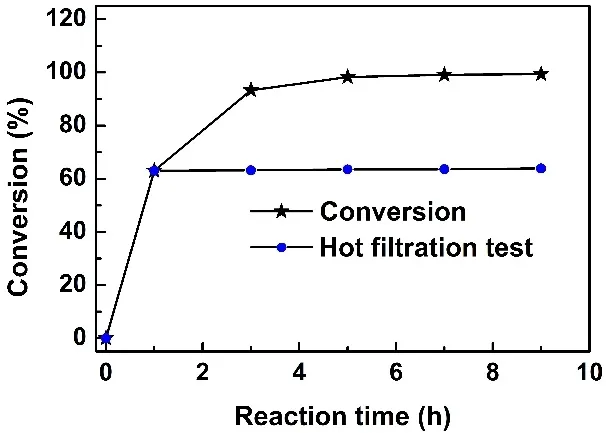
图10 Cu2-MnOx热过滤测试Fig. 10 Hot filtration test of Cu2-MnOx.Reaction conditions: THQL (0.25 mmol), catalyst (25 mg),DMF (5.0 mL), 130 °C, air balloon.
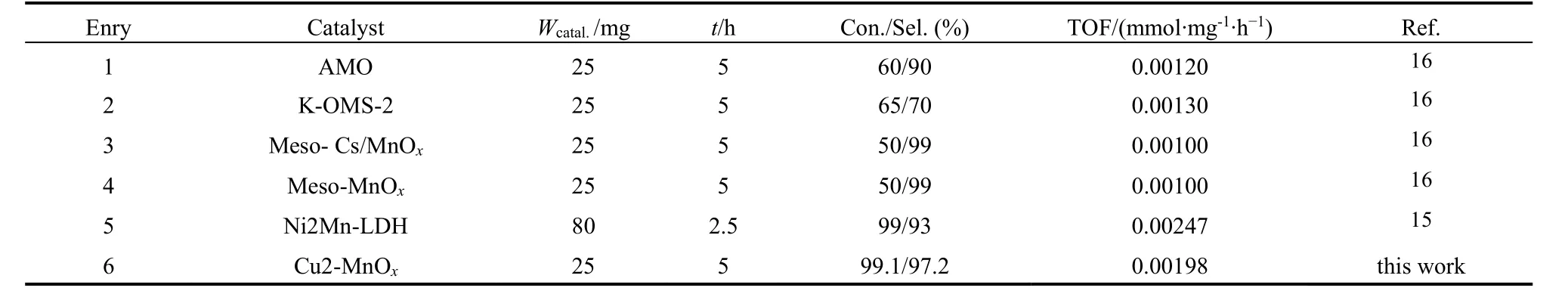
表5 不同锰基多相催化剂对THQL合成QL催化性能比较Table 5 Comparison of catalytic performance of various manganese-based heterogeneous catalyst for the synthesis of QL from THQL.
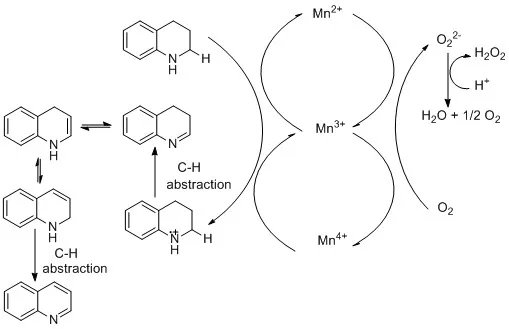
图11 THQL氧化芳构化可能的机理Fig. 11 Proposed mechanism of oxidative aromatization of THQL.
根据文献报道的结果15,16,我们假设THQL的氧化脱氢芳构化可能的机理如图11。脱氢是从氮原子转移到Mn4+或Mn3+单个电子引发的,然后形成的胺基自由基进行了α-C——H的脱除得到亚胺中间体,同时伴随着水的形成和还原锰的再氧化,亚胺中间体和烯胺存在平衡。再一次单电子转移从富电子的烯胺氮原子开始,然后进行α-C——H的第二次脱除得到QL。在催化循环中,胺自由基的形成导致表面活性锰中心的还原,而锰中心的还原又导致晶格氧的释放。因此,在锰氧化物催化的氧化反应机理中,锰氧化物的晶格氧流动性是其重要性质37。从表4 (Entry5)可以看出CuO自身的催化活性很低,但CuO的掺杂可以促进无定型MnOx晶格氧的流动性,这也得到H2-TPR结果证实。Mn3+在氧化脱氢中具有重要作用,是关键的催化活性位15,38。CuO的掺杂也提高了Mn3+的比例,这也是掺杂后无定型锰氧化物具有高催化活性的重要因素。
4 结论
我们采用共沉淀法(草酸路线)制备了高效的CuO掺杂的无定型MnOx催化剂,并应用于THQL氧化脱氢芳构化。少量CuO的掺杂极大的提高了MnOx的催化性能,当Cu的掺杂量为0.81% (w)时,Cu2-MnOx具有理想的转化率(99.1%)和选择性(97.2%),TOF值达0.00198 mmol·mg-1·h-1。在无定型的锰氧化物中,CuO和MnOx的协同作用导致Cu2-MnOx具有增大的孔径、较低的还原温度、最高的Mn3+和吸附氧含量及最高的Mn3+/Mn4+,这是催化性能提高的关键因素。这种体系采用非常廉价的非贵金属,催化剂制备工艺简单,用空气作为唯一的氧化剂,没有配体和碱的加入,反应条件温和,催化剂可多次重复使用,是一种非常有希望工业化推广的绿色化工过程。
