犬细小病毒河南株的分离鉴定及全基因组序列分析
2019-05-28孙浩杰张百惠蔡亚南魏战勇
唐 磊,张 弛,秦 亚,孙浩杰,张 楠,张百惠,蔡亚南,魏战勇
(1.吉林农业大学,吉林 长春 130118; 2.河南农业大学,河南 郑州 450002)
犬细小病毒病是由犬细小病毒(Canine parvovirus,CPV)引起的一种急性、高度接触性传染病,以出血性肠炎和心肌炎为主要发病特征,并伴有剧烈呕吐、便血、食欲不振、精神沉郁等临床症状。该病毒于1978年在澳大利亚患肠炎的犬中首次被分离出[1],随后在世界范围内陆续被报道。1983年,徐汉坤等[2]在我国首次正式报道了该病的流行情况。犬细小病毒病与犬瘟热、犬冠状病毒病并称为犬类的三大传染病,严重危害到我国养犬业与宠物行业的健康发展。
CPV为无囊膜、单链DNA病毒。病毒基因组由5 323个碱基组成,含有2个主要的开放性阅读框(Open reading frame,ORF),分别编码2个非结构蛋白NS1和NS2与3个结构蛋白VP1、VP2和VP3[3]。病毒衣壳由60个结构蛋白组成,其中VP2是病毒衣壳中最主要的结构蛋白,占病毒衣壳的90%,有53~54个。在VP2蛋白的N端以及蛋白转角区loop1和loop3均存在重要的抗原表位,因此,VP2基因的序列发生氨基酸替代,可能引起病毒抗原性的改变[4]。VP2基因组两端存在2个发卡结构(3′端Y型和5′端U型),在病毒复制过程中起着重要的作用,但这2个发卡结构也能够阻碍病毒基因组的全序列测序及分析[5]。
在本研究中,将从经CPV胶体金试纸鉴定为阳性的病犬粪便中成功分离出的1株CPV病毒,进行理化性质、血凝试验、病毒全基因组测序鉴定以及基因组序列分析,以期了解河南省目前CPV流行趋势,为该病的治疗与预防提供参考和依据。
1 材料和方法
1.1 试验材料
1.1.1 供试细胞与试剂 胎牛血清、MEM细胞培养液购自Hyclone公司;蛋白酶K、琼脂糖购自Promega公司;DL2000 DNA Marker、pMD18-T载体购自大连宝生物公司;DNA凝胶回收试剂盒、DH5α感受态细胞购自北京康为世纪生物科技有限公司;质粒提取试剂盒购自OMEGA公司;DNA提取试剂盒、PrimeSTAR HS DNA Polymerase购自上海生工生物工程有限公司;其他常规试剂均为分析纯。F81细胞为河南省动物性食品安全重点实验室保存。
1.1.2 病料的收集与处理 于郑州市某动物医院,收集经CPV胶体金试纸鉴定为阳性的病犬血便病料1份。将收集的CPV感染犬的血便病料,按照1∶10的比例用PBS稀释,加入等量的氯仿混匀,再加入双抗,于4 ℃过夜。然后于8 000 r/min离心10 min,取上清液,经0.22 μm滤器过滤除菌,-80 ℃保存备用。
1.2 试验方法
1.2.1 病料DNA提取 取处理好的病料样品提取DNA,具体步骤参照DNA提取试剂盒的操作说明,-40 ℃保存备用。
1.2.2 检测引物PCR鉴定 根据GenBank收录的CPV-LZ2序列(登录号:JQ268284),针对病毒的NS1、VP2基因区域,运用Primer 5.0软件设计2对检测引物(表1)[6],引物由武汉奥科鼎盛生物科技有限公司合成。
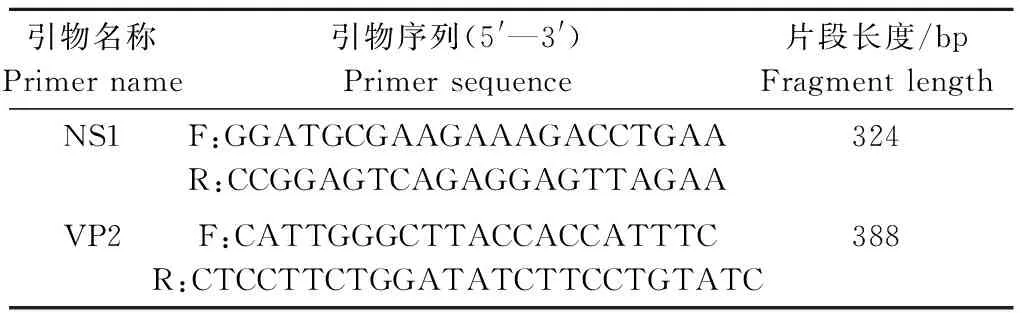
表1 CPV检测引物序列
采用50 μL的PCR扩增体系:TaqMix 25 μL,上、下游引物各2 μL,模板DNA 4 μL,最后用灭菌双蒸水添加至50 μL。反应条件:94 ℃ 5 min;94 ℃ 30 s,60 ℃ 30 s,72 ℃ 40 s,35个循环;72 ℃ 10 min,4 ℃保存。取8 μL PCR产物于1%琼脂糖凝胶中进行电泳鉴定,并用DNA凝胶回收试剂盒进行回收与纯化。将纯化后的产物连接到pMD18-T载体上,转化至DH5α感受态细胞,涂于Amp+琼脂平板上,于37 ℃培养箱培养过夜。挑选单个菌落进行扩大培养,将菌液PCR鉴定为阳性的菌液送往武汉奥科鼎盛生物科技有限公司测序。
1.2.3 CPV病毒分离 取1.1.3中处理好的病料样品,进行10倍稀释后同步接种于F81细胞[7],另设有不接种的F81细胞作为阴性对照,于37 ℃细胞培养箱中培养。每日观察细胞病变效应(Cytopathic effect,CPE)并进行记录,待CPE达到80%时,收取病毒,并盲传5代,取第5代病毒[8],运用Reed-Muench法测定其组织半数感染量(Tissue culture infective dose,TCID50)[9]。收获每代病毒,-80 ℃保存。
1.2.4 CPV血凝(HA)试验 根据CPV可以凝集猪红细胞这一特性,制备1%猪红细胞悬液,取第5代病毒,按血凝试验[10]的操作方法鉴定CPV病毒的血凝效价。
1.2.5 CPV理化性质试验 用20%乙醚处理CPV病毒后进行脂溶剂敏感试验;用pH值为3.0的酸处理病毒进行耐酸性试验;56 ℃加热30 min,对病毒进行耐热性试验[11]。每组试验均设立阴性对照组,进行相应处理后,分别测定阴性对照组与试验组的TCID50。
1.2.6 CPV全基因组测序 根据GenBank收录的CPV-LZ2序列(登录号:JQ268284),运用Primer 5.0软件设计9对引物序列(表2)。其中,Y1、Y2用于Y型发卡结构扩增,U1、U2用于U型发卡结构扩增,A1—A5用于编码区序列扩增,引物均由武汉奥科鼎盛生物科技有限公司合成。

表2 CPV全基因组引物序列Tab.2 CPV whole genome primers
编码区片段扩增采用50 μL的PCR扩增体系:TaqMix 25 μL,上、下游引物各2 μL,模板DNA 4 μL,最后用灭菌双蒸水添加至50 μL。反应条件:94 ℃ 5 min;94 ℃ 30 s,60 ℃ 30 s,72 ℃ 40 s,35个循环;72 ℃ 10 min,4 ℃保存。取PCR产物8 μL于1%琼脂糖凝胶中进行电泳鉴定,并用DNA凝胶回收试剂盒进行回收与纯化。两端的发卡结构片段扩增采用25 μL反应体系:模板DNA 2 μL,上游引物1 μL,dNTP 2 μL,5×GC Buffer 5 μL,混匀后于98 ℃反应5 min,迅速置于冰上5 min,然后加入PrimeSTAR HS DNA Polymerase 0.5 μL,混匀后于72 ℃反应3 min,加入下游引物1 μL,添加灭菌双蒸水至25 μL。反应条件:98 ℃ 45 s;98 ℃ 30 s,60 ℃ 30 s,72 ℃ 30 s,35个循环;72 ℃ 10 min,4 ℃保存。取PCR产物8 μL于1%琼脂糖凝胶中进行电泳鉴定,并用DNA凝胶回收试剂盒进行回收与纯化。
将纯化后的产物连接到pMD18-T载体上,转化至DH5α感受态细胞,涂于Amp+琼脂平板上,于37 ℃培养箱培养过夜。挑选单个菌落进行扩大培养,将菌液PCR鉴定为阳性的菌液送往武汉奥科鼎盛生物科技有限公司测序。
1.2.7 CPV全基因组序列分析 将1.2.6中引物的扩增片段用DNAStar软件进行序列拼接,运用MegAlign软件将拼接好的CPV全基因组序列与GenBank上收录的17株国内CPV参考毒株(表3)进行核苷酸同源性比对分析,并绘制系统进化树。
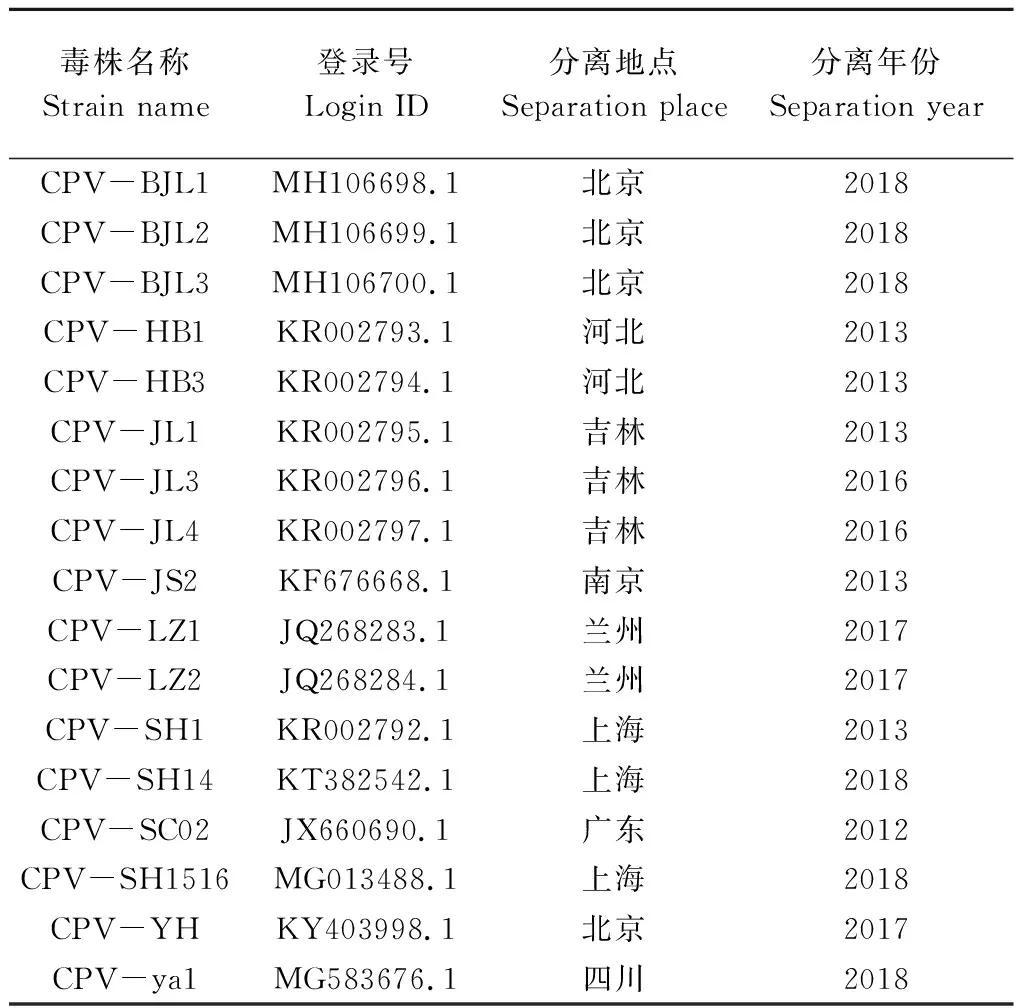
表3 CPV参考毒株信息
2 结果与分析
2.1 目的片段的PCR鉴定结果
引物NS1和VP2 PCR扩增片段经1%琼脂糖凝胶电泳检测后,分别得到324 bp和388 bp的核酸电泳条带(图1),与预期值大小相符。并将阳性菌液的测序结果与GenBank收录的CPV-LZ2序列(登录号:JQ268284)参考株序列进行比对,结果表明收集的血便病料内存在CPV。
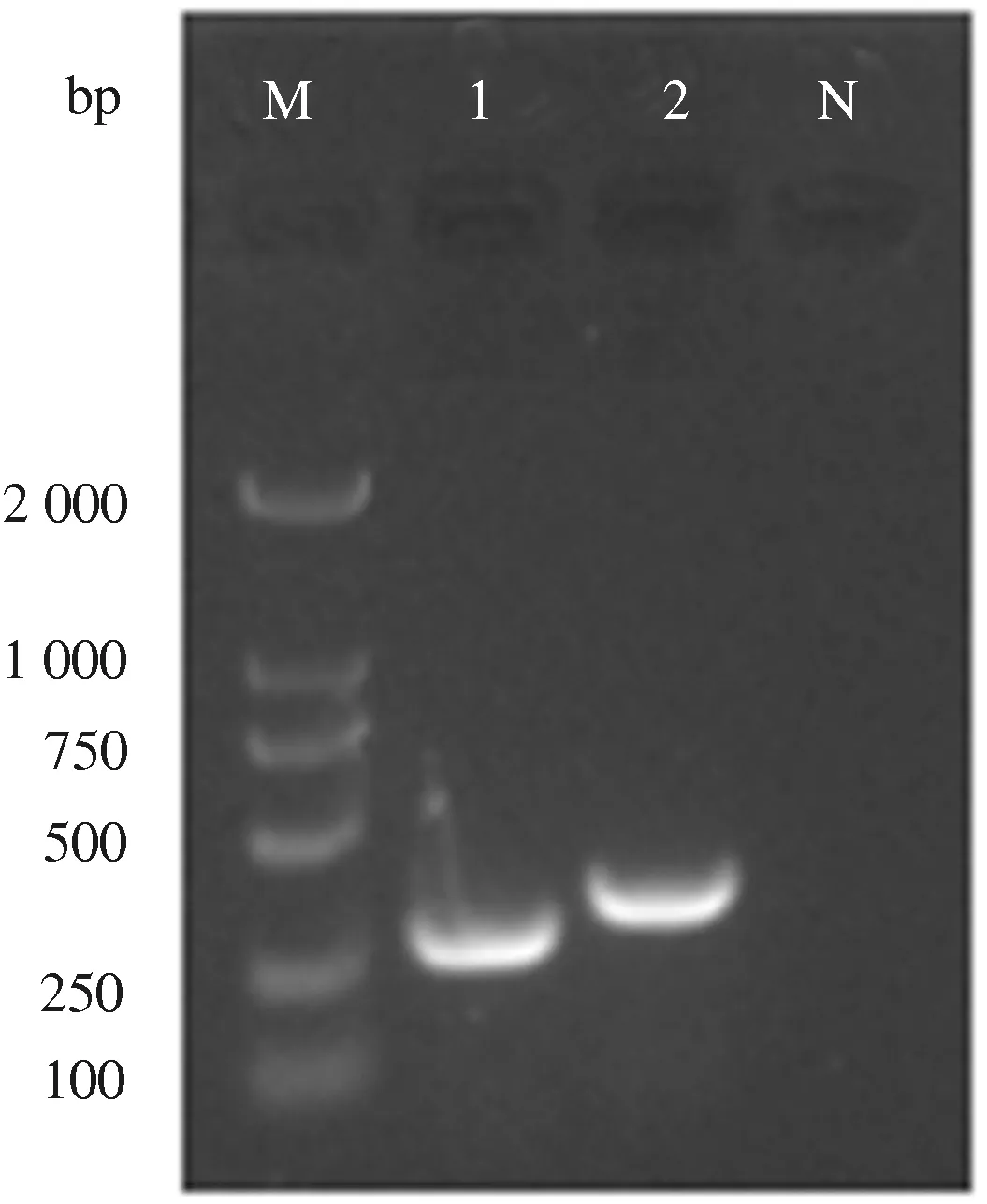
M:DL2000 DNA Marker; 1:引物NS1扩增片段; 2:引物VP2扩增目的片段; N:阴性对照 M:DL2000 DNA Marker; 1:Primer NS1 amplified fragment;2:Primer VP2 amplified target fragment; N:Negative control
2.2 CPV病毒的分离
接种10倍稀释的血便病料样品于F81细胞后,经过48 h盲传至第2代时,F81细胞开始出现脱落、拉网、聚集、变形等明显的细胞病变,继续盲传至第5代,F81细胞均产生典型的CPE(图2A),而阴性对照组未接种病料样品的F81细胞呈梭型,生长良好,形态正常,细胞连接紧密,未出现任何细胞病变(图2B)。收获每代病毒,-80 ℃保存,将其命名为CPV/HN1。
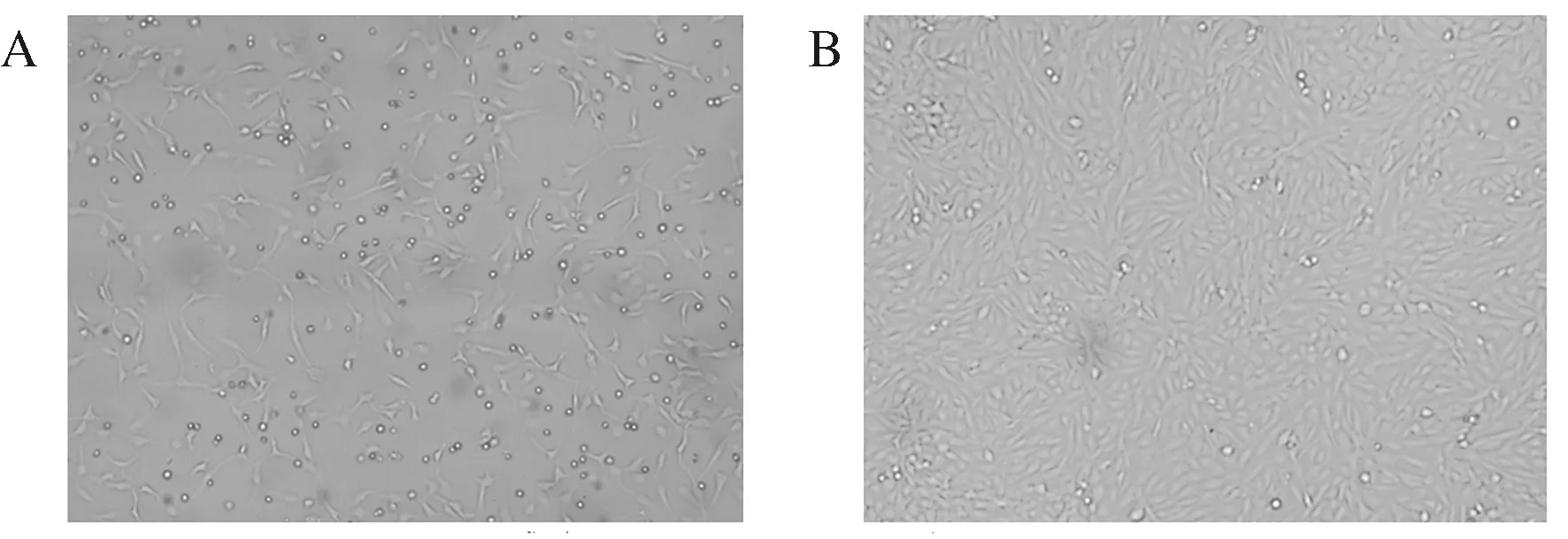
A:CPV感染F81细胞; B:正常F81细胞
2.3 CPV/HN1病毒的血凝效价检测结果
待CPV/HN1病毒盲传至第5代,取第5代病毒。根据犬细小病毒可以对猪红细胞产生凝集效应,制备1%猪红细胞悬液,按HA试验操作方法进行血凝效价测定,结果显示其血凝效价为1∶256。
2.4 CPV/HN1病毒TCID50的测定结果
按TCID50试验操作方法对盲传至第5代的CPV/HN1病毒进行TCID50测定,根据Reed-Muench法计算CPV/HN1第5代病毒的TCID50,其值为10-4.85/0.1 mL。
2.5 CPV/HN1病毒理化性质试验结果
经乙醚和酸(pH值3.0)加热处理后,CPV/HN1病毒的TCID50均无明显变化,处理组与对应的未处理对照组TCID50均无显著差异,说明该病毒对酸、热和有机溶剂均具有较强抵抗力,这与犬细小病毒相关理化性质相符。
2.6 CPV/HN1病毒全基因组测序结果及序列分析
如图3所示,CPV/HN1病毒编码区PCR扩增片段经1%琼脂糖凝胶电泳检测,结果显示,A1的目的片段长度为1 156 bp、A2的目的片段长度为1 004 bp、A3的目的片段长度为1 052 bp、A4的目的片段长度为1 061 bp、A5的目的片段长度为1 016 bp,均与预期结果相符;如图4所示,经PCR扩增后,Y1的目的片段长度为66 bp、Y2的目的片段长度为712 bp、U1的目的片段长度为312 bp、U2的目的片段长度为101 bp,与预期结果相符。
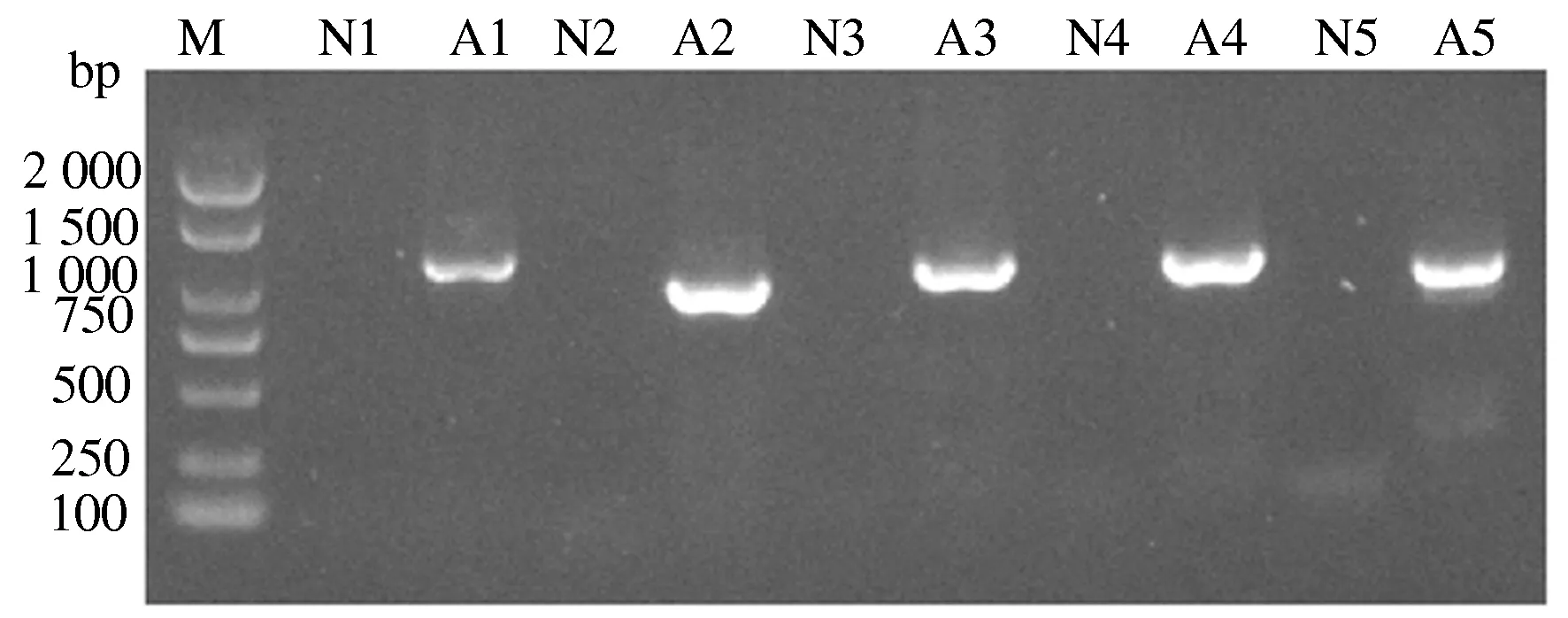
M:DL2000 DNA Marker; N1—N5:阴性对照;A1—A5:编码区域目的片段扩增
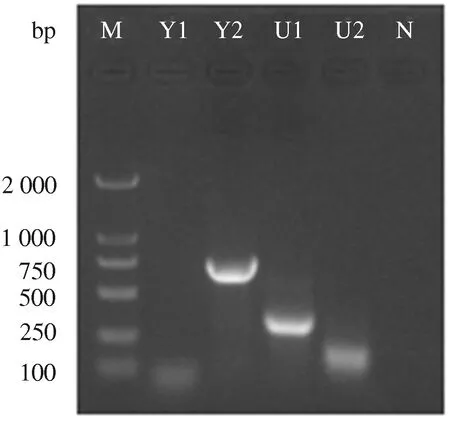
M:DL2000 DNA Marker; Y1—U2:发卡结构目的片段扩增; N:阴性对照
如图5所示,对CPV/HN1病毒的VP2氨基酸序列进行分析发现,其发生了Asn426Asp的氨基酸突变,但并没有发生Val555Ile的氨基酸突变,同时还发生了Ser297Ala的突变,证明其属于New CPV-2b亚型。拼接得到全基因组序列总长为5 052 bp,与模板序列进行比对,其中U型发卡结构全长为188 bp,Y型发卡结构全长为120 bp,ORF1全长为2 007 bp并编码非结构蛋白NS1,ORF2全长为2 257 bp并编码结构蛋白VP1、VP2。
如图6所示,通过与GenBank上登录的国内17株CPV核苷酸序列进行比对,核苷酸同源性在96.4%~99.9%,其中CPV/HN1病毒与广东分离株CPV-SC02/JX660690.1/2012同源性最低为96.4%,与北京分离株CPV-BJL1/MH106698.1/2018和CPV-BJL3/MH106700.1/2018同源性最高为99.9%。此外,CPV/HN1病毒同2018年以来北京的3株分离株具有较高的同源性,因此推测CPV/HN1病毒与国内北京分离毒株具有相同的起源。
CPV全基因组进化树分析如图7所示,CPV/HN1病毒与北京毒株CPV-BJL1/MH106698.1/2018、CPV-BJL2/MH106699.1/2018、CPV-BJL3/MH106700.1/2018处于同一分支且具有较近的亲缘关系,与国内兰州、吉林、上海等分离株亲缘关系较远。总体而言,CPV/HN1病毒分离株与近年来北京分离株亲缘关系较近,同国内其他分离株亲缘关系疏远,推测CPV/HN1病毒分离株的流行同北京毒株相关。

图5 CPV/HN1病毒VP2蛋白氨基酸序列分析Fig.5 Amino acid sequence analysis of VP2 protein of CPV/HN1
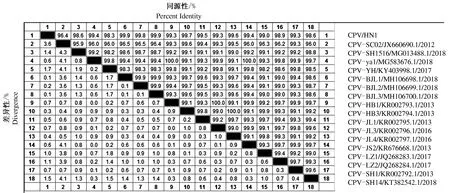
图6 CPV/HN1病毒全基因组核苷酸同源性分析Fig.6 Nucleotide homology analysis of CPV/HN1 genome
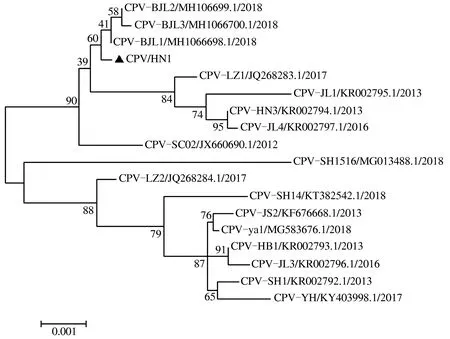
图7 CPV/HN1病毒进化树分析Fig.7 Analysis of CPV/HN1 phylogenetic tree
3 结论与讨论
随着人们生活水平的提高,以宠物行业为首的养犬行业高速发展。随着养犬数量日益增加,CPV发病率不断上升,严重制约了我国养犬业的健康发展,每年造成巨大的经济损失[12]。因此,加强CPV感染机制及流行病学调查的研究,对该病的预防及治疗具有重要的生产意义。
目前,CPV的预防主要以免疫接种为主,但即使进行有效的疫苗接种,该病依然在世界范围内广泛流行[13]。自1978年该病毒首次被分离以来,病毒先后进行了多次基因变异,产生了3种主要的抗原变异型CPV-2a、CPV-2b和CPV-2c,其中变异型CPV-2a和CPV-2b主要在一些亚洲国家流行,变异型CPV-2c主要分布于欧洲及美洲各国。近年来研究发现,新的变异型New CPV-2a、New CPV-2b和New CPV-2c在亚洲、美洲和欧洲等地开始出现[14]。有研究发现,VP2基因的定向漂移引发了各种变异型的产生,CPV-2a的VP2蛋白相较于CPV-2,发生了Met87Leu、Ile101Thr、Ala300Gly、Asp305Tyr以及Val555Ile的氨基酸突变;而CPV-2b的VP2蛋白相较于CPV-2a多了Asn426Asp的氨基酸突变,但并没有发生Val555Ile的氨基酸突变;CPV-2c的VP2蛋白相较于CPV-2b除发生了相同的氨基酸突变外,还在Asp426Glu和Ser297Ala发生了突变,这些突变导致了生物学性质的改变[15]。通过与CPV-2a、CPV-2b比较发现,新变异株New CPV-2a、New CPV-2b发生了Ser297Ala的氨基酸突变,该位点的氨基酸突变可作为判断New CPV-2a和New CPV-2b的标志,另外,某些New CPV-2a和New CPV-2b还发生了Phe267Tyr、Tyr324Ile和Thr440Ala突变[15]。CPV虽然属于DNA病毒,但是它的复制过程依然依赖于宿主细胞,并且由于它属于单链DNA,在复制过程中的复制效率及纠错能力不高,导致该病毒基因组的碱基突变率高于一般的DNA病毒,而与某些RNA病毒相当,但该特点导致CPV对环境与宿主的适应性及逃避宿主免疫应答能力大大提高,从而提升了CPV的进化效率以及在宿主动物中的传播能力[16]。
CPV全基因组5′端和3′端存在有末端回文序列形成的末端发卡结构,在病毒的复制过程中起着起始和包装信号等重要作用[17]。目前为止,由于其两端发卡结构的克隆与测序的困难性,导致基因组数据库中仅仅存在几株完整的CPV全基因组序列,这严重阻碍了CPV全基因组及其感染机制的研究工作[18]。结合文献报道,本研究最终克隆并测序出末端2个发卡结构,成功拼接出完整的CPV全基因组序列。
本研究从患病犬血便中成功分离出1株CPV毒株,通过对该CPV病毒分离株的PCR鉴定、血凝试验、病毒分离试验和病毒理化性质试验,以及全基因组测序,证明该毒株确实是CPV,并将其命名为CPV/HN1。对该毒株的全基因组克隆及测序显示,该毒株为New CPV-2b亚型,通过与17株国内CPV流行毒株全基因组序列核苷酸同源性分析,显示同源性在96.4%~99.9%,相较于国内其他毒株,与北京毒株,同源性较高,其中与CPV-BJL1/MH106698.1/2018和CPV-BJL3/MH106700.1/2018北京分离株同源性最高为99.9%。CPV/HN1病毒的成功分离,为河南省CPV的流行、遗传进化等研究提供理论数据基础,为CPV疫苗的开发与应用提供材料基础,同时为CPV的感染机制及感染模型建立研究提供选择的方向。
