Design,synthesis and biological evaluation of selective survivin inhibitors
2019-05-17MinXiaoYiXueZhongzhiWuZiNingLeiJinWangZheShengChenWeiLi
Min Xiao,Yi Xue,Zhongzhi Wu,Zi-Ning Lei,Jin Wang,Zhe-Sheng Chen,Wei Li,✉
1 Department of Pharmaceutical Sciences,University of Tennessee Health Science Center,Memphis,TN,38163,USA;
2 Department of Pharmaceutical Sciences,College of Pharmacy and Health Sciences,St.John’s University,Queens,NY 11439,USA.
AbstractThe differential distribution between cancer cells and normal adult tissuesmakessurvivin avery attractivecancer drug target.We have previously reported a series of novel selective survivin inhibitors with the most potent compound MX106 reaching nanomolar activity in several cancer cell lines.Further optimization of the MX106 scaffold leadsto thediscovery of morepotent and moreselectivesurvivin inhibitors.Variousstructural modi f i cations weresynthesized and their anticancer activitieswereevaluated to determinethestructureactivity relationshipsfor this MX106 scaffold.In vitro anti-proliferative assays using two human melanoma cell lines showed that several new analogs have improved potency compared to MX106.Very interestingly,these new analogs generally showed signi f i cantly higher potency against P-glycoprotein overexpressed cells compared with the corresponding parental cells,suggesting that these compounds may strongly sensitize tumors that have high expressions of the Pglycoprotein drug ef f l ux pumps.Western blotting analysis con f i rmed that the new MX106 analogs maintained their mechanism of actions by selectively suppressing survivin expression level among major inhibitors of apoptotic proteins and induced strong apoptosis in melanoma tumor cells.
Keywords:selective survivin inhibitors,structure activity relationships,melanoma,human epidermoid carcinoma,colorectal cancer,P-glycoprotein drug ef f l ux pumps
Introduction
Survivin is the smallest(molecular weight 16.5 kDa)member of the IAP(Inhibitor of Apoptosis Protein)family[1-2].It is a unique anti-tumor therapy target because:(a)it acts as an essential anti-apoptosis guard which protects cells from the apoptotic cascade by binding to the activated caspases and neutralizing proapoptotic receptor[3-5];(b)itisakey regulator of mitosis asacomponent of thechromosomal passenger complex(CPC)[6-7];(c)it widely participates in tumorigenesis signaling pathways such as Akt,p53,Wnt-2 and MDM2[8-10];(d)it is highly expressed in most types of human cancer cells and embryonic tissues where rapid cell growth isneeded,buthasvery low expression in normal adult differentiated tissues[11-12];(e)its expression level is closely correlated with tumor metastasis,poor disease prognosis,and high risk of chemo/radio-resistance[8,11,13-17].
However,due to the nature of survivin function(protein-protein interaction)and its cell cycle dependent expression,the existing armory of survivin inhibitors is still very limited[8,18].Most current small molecular survivin inhibitors do not directly bind to the survivin protein,but rather interfere with survivin production(gene transcription,post-translation modi f ication)or accelerate its degradation[8,12,18-20].
Multidrug resistance(MDR)remains a major obstacle in cancer chemotherapy,accounting for more than 90%failure in clinical treatment to cancer patients.A family of ATP binding cassette(ABC)transporters,which isaclassof drug ef f l ux pumps,playsakey rolein developing MDR[21].At least 15 human ABC transporters,such as P-glycoprotein(P-gp),multidrug resistant protein1(MRP1)and breast cancer resistant protein(BCRP)have been found to mediate MDR by inducing drug ef f l ux[22].Among them,P-gp has been themost extensively identi f i ed MDRin cancer cellsand a broad range of chemotherapy drugs are known to be substratestransported by P-gp.Asthemostpotentsmall molecule survivin inhibitor reported so far,YM155 is a survivin gene promotor inhibitor and is well known to be highly susceptible to clinically relevant MDR,including but not limited to P-gp overexpression[18-19,23-24].It also showspotential toxicities,which calls for special dose administration arrangements.Therefore,it is important to develop novel selective survivin inhibitorsthat can overcomeclinically relevant MDR.
Utilizing high throughput virtual screening and biological test oriented strategy,our laboratory has previously developed aplatformof cell permeablesmall molecular survivin inhibitors,which selectively and effectively reduced survivin expression level in human melanoma and prostate cancer cells,with potent antitumor growth ef f i cacy as validated by a human melanoma xenograft model[25-26].UC112 is the lead compound we identi f i ed through virtual screening and in vitro biological studies.Preliminary optimization of UC112 leads to the discovery of a potent and selective survivin inhibitor,MX106.Thestructureof UC112 and MX106 areshown in Fig.1.In thisreport,wedescribed our efforts to further optimize the MX106 scaffolds which lead to the discovery of more potent MX106 analogs for selective survivin inhibition in tumor cells.We report that these new analogs show strong antiproliferative potency against human melanoma and colorectal cancer cells,effectively overcome P-gp mediated drug resistance,and maintain their mechanisms of action by selectively inhibiting survivin expression and inducing cancer cell apoptosis.Structure-activity relationships(SAR)are determined for these new analogs to support further development of this unique scaffold as a potential anticancer agent.
Materials and methods
General
All reagents were purchased from Sigma-Aldrich Chemical Co.,Alfa Aesar(Ward Hill,MA),and AK Scienti f i c(Mountain View,CA)and were used without further puri f i cation.Routine thin layer chromatography(TLC)was performed on aluminum-backed Uniplates(Analtech,Newark,DE).NMR spectra were obtained on a Varian Inova-500 spectrometer(Agilent Technologies,Santa Clara,CA)or a Bruker Ascend 400(Billerica,MA)spectrometer.Chemical shifts are reported as parts per million(ppm)relative to TMS in CDCl3.High resolution mass spectra were collected in positive detection mode on a Waters Xevo G2-S Tof instrument equipped with an electron-spray ionization(ESI)source(Milford,MA).
Synthesis
Preparation of 5-chloromethyl-8-quinolinol hydrochloride(2)
A mixture of 5.84 g(40.0 mmol)of 8-quinolinol,50 ml of concentrated hydrochloric acid,and 6.4 ml of 37%formaldehyde was treated with 0.6 g of zinc chloride and stirred for 12 hours.The mixture was f i ltered,washed with copious acetone and dried to give compound 2 as a yellow solid(7.2 g,78%).1H NMR(400 MHz,deuterium oxide)δ9.12(dd,J=8.7,1.4 Hz,1H),8.88(dd,J=5.5,1.4 Hz,1H),7.97(dd,J=8.7,5.4 Hz,1H),7.59(d,J=8.0 Hz,1H),7.24(d,J=7.9 Hz,1H),4.93(s,2H).
General procedure for the synthesis of compounds(3a-c)
To a solution of substituted benzyl alcohol 3(6 mmol)in anhydrous THF(30 mL)was added sodium hydride(60%dispersion in mineral oil,0.72 g,18 mmol)at 0°C.The suspension was stirred at 0°Cfor 30 minutes.Salt 2(1.15 g,5mmol)was added to the suspension.The mixture was stirred at r.t for 3 hours.Water was added to the suspension,and the mixture became homogeneous.The mixture was extracted by ethyl acetate and washed with brine,dried over anhydrous sodium sulfate and concentrated to get the crude.The crude compound was puri f i ed by f l ash chromatography(ethyl acetate:hexane 1:3)
5-(((4-azidobenzyl)oxy)methyl)quinolin-8-ol(3a)
1HNMR(400 MHz,CDCl3)δ8.78(d,J=2.8 Hz,1H),8.41(d,J=8.4 Hz,1H),7.45(d,J=8.0 Hz,2H),7.37(d,J=7.2 Hz,1H),7.24(d,J=8.0 Hz,2H),7.11(d,J=7.2 Hz,1H),4.84(s,2H),4.52(s,2H).
5-(((4-ethynylbenzyl)oxy)methyl)quinolin-8-ol(3b)
1HNMR(400 MHz,CDCl3)δ8.77(d,J=2.8 Hz,1H),8.44(d,J=8.4 Hz,1H),7.45(d,J=8.0 Hz,2H),7.38(d,J=7.2 Hz,1H),7.25(d,J=8.0 Hz,2H),7.10(d,J=7.2 Hz,1H),4.84(s,2H),4.50(s,2H),3.07(s,3H).
5-(((2-bromo-4-methylbenzyl)oxy)methyl)quinolin-8-ol(3c)
1H NMR(400 MHz,Chloroform-d)δ8.85(dd,J=4.3,1.6 Hz,1H),8.54(dd,J=8.5,1.5 Hz,1H),7.60-7.51(m,2H),7.46(d,J=7.8 Hz,1H),7.23(d,J=7.7 Hz,1H),7.17(td,J=5.0,2.5 Hz,2H),4.89(s,2H),4.50(s,2H),2.41(s,3H).
General procedure for the synthesis of compounds(4a-c)
An equimolar mixture of the substrates 3,paraformaldehyde,and the pyrrolidine in anhydrous ethanol(30 mL)was re f l uxed for 4 hours under argon.After cooling,the solvent was evaporated under reduced pressure.The crude compound was puri f i ed by f l ash chromatography(Dichloromethane:methanol 20:1).
5-(((4-azidobenzyl)oxy)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol(4a)
1HNMR(400 MHz,CDCl3)δ8.89(dd,J=4,1.6 Hz,1H),8.37(dd,J=8.4,1.6 Hz,1H),7.41(dd,J=8.4,4.0 Hz,1H),7.31(d,J=8.4 Hz,2H),7.21(s,1H),7.00(d,J=8.4 Hz,2H),4.84(s,2H),4.53(s,2H),3.98(S,2H),2.70(m,4H),1.88(m,4H).HRMS(ESI):m/z calculated for C22H23N5O2+ H+[M+ H]+:390.1930;Found:390.1942.
5-(((4-ethynylbenzyl)oxy)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol(4b)
1HNMR(400 MHz,CDCl3)δ8.88(dd,J=8.4,1.6 Hz,1H),8.37(dd,J=8.4,1.6 Hz,1H),7.46(d,J=8.4 Hz,2H),7.41(dd,J=8.4,4.0 Hz,1H),7.28(d,J=8.4 Hz,2H),7.21(s,1H),4.84(s,2H),4.55(s,2H),3.99(s,2H),3.07(s,1H),2.72-2.69(m,4H),1.89-1.86(m,4H).HRMS(ESI):m/z calculated for C24H24N2O2+H+[M+H]+:373.1916;Found:373.1924.
5-(((2-bromo-4-methylbenzyl)oxy)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol(4c)
1H NMR(400 MHz,Chloroform-d)δ8.79(d,J=4.1 Hz,1H),8.43-8.22(m,1H),7.47-7.29(m,3H),7.16-7.04(m,2H),4.78(s,2H),4.43(s,2H),4.08(s,2H),2.96-2.70(m,4H),2.31(s,3H),1.97-1.76(m,4H).Exact mass for C23H25BrN2O2:442.1079;HRMS:[M+H]+:443.1170
Preparation of 5-((naphthalen-2-ylmethoxy)methyl)quinolin-8-ol(5)
To a solution of the alcohol in anhydrous DMF(10 mL)was added sodium hydride(60%dispersion in mineral oil,0.72 g,18 mmol)at 0°C.The suspension was stirred at 0°C for 30 minutes.Salt 2(1.15 g,5 mmol)was added to the suspension.The mixture was stirred at r.t for 3 hours.Water was added to the suspension,the mixture became homogeneous.The mixturewasextracted by ethyl acetateand washed with brine,dried over anhydrous sodium sulfate and concentrated to get the crude.The crude compound was puri f i ed by f l ash chromatography(ethyl acetate:hexane 1:3)1H NMR(400 MHz,Chloroform-d)δ8.77(dd,J=4.2,1.6 Hz,1H),8.38(dd,J=8.5,1.6 Hz,1H),8.07-8.00(m,1H),7.92-7.77(m,2H),7.52-7.40(m,5H),7.36(dd,J=8.5,4.2 Hz,1H),7.11(d,J=7.7 Hz,1H),4.99(s,2H),4.90(s,2H).
Preparation of 5-((naphthalen-2-ylmethoxy)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol(6)
An equimolar mixture of the substrates 5,paraformaldehyde,and the pyrrolidine in anhydrous ethanol(30 mL)was re f l uxed for 4 hours under argon.After cooling,the solvent was evaporated under reduced pressure.The crude compound was puri f i ed by f l ash chromatography(Dichloromethane:methanol 20:1).1H NMR(400 MHz,Chloroform-d)δ8.85(dd,J=4.2,1.7 Hz,1H),8.30(dd,J=8.5,1.6 Hz,1H),8.12-7.98(m,1H),7.92-7.77(m,2H),7.53-7.40(m,5H),7.35-7.28(m,2H),5.02(s,2H),4.90(s,2H),4.04(s,2H),2.87-2.67(m,4H),1.90(p,J=3.2 Hz,4H).Exact mass for C26H26N2O2:398.1994;HRMS:[M+H]+:399.2071
Preparation of 5-(morpholinomethyl)quinolin-8-ol(7)
To a solution of morpholine(6 mmol)in THF was added salt2(2 mmol).Themixturewasre f l uxed for 3hr.The white precipitate was f i ltered,the f i ltrate was evaporated.The crude was puri f i ed by f l ash chromatography to produce 7.1H NMR(400 MHz,chloroformd)δ8.79(dd,J=4.2,1.6 Hz,1H),8.67(dd,J=8.5,1.6 Hz,1H),7.47(dd,J=8.5,4.2 Hz,1H),7.33(d,J=7.7 Hz,1H),7.07(d,J=7.7 Hz,1H),3.79(s,2H),3.70-3.61(m,4H),2.45(dd,J=5.6,3.7 Hz,4H).
Preparation of 5-(morpholinomethyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol(8)
An equimolar mixture of the substrates 7,paraformaldehyde,and the pyrrolidine in anhydrous ethanol(30 mL)was re f l uxed for 4 hours under argon.After cooling,the solvent was evaporated under reduced pressure.The crude compound was puri f i ed by f l ash chromatography(Dichloromethane:methanol 20:1).1H NMR(400 MHz,Chloroform-d)δ8.86(ddd,J=5.9,4.1,1.6 Hz,1H),8.60(td,J=8.5,1.7 Hz,1H),7.40(dt,J=8.4,4.2 Hz,1H),7.19(d,J=29.0 Hz,1H),3.95(d,J=31.1 Hz,2H),3.86(s,1H),3.81-3.74(m,3H),3.66(t,J=4.7 Hz,2H),2.76-2.58(m,4H),2.57-2.40(m,4H),1.95-1.69(m,4H).Exact mass for C19H25N3O2:327.1947;HRMS:[M+H]+:328.2026
Preparation of 2-chloroquinolin-8-ol(10)
Quinoline-2,8-diol(5.30 g,32.89 mmol)was suspended in anhydrous DMF(30 mL).Thionyl chloride(9.45 mL,131.50 mmol)was added dropwise via a syringe at 0°C under argon.The resulted solution was stirred and heated to 50°C for 4 hours.Then,the reaction was quenched by pouring the solution into ice water followed by extraction with ethyl acetate(3×50 mL),washed with brine(2×50 mL),dried over anhydrous MgSO4and concentrated under reduced pressure and puri f i ed by silica gel column chromatography(eluting with CH2Cl2)to giveapaleyellow solid product,5.0 g,84.7%yield.
Preparation of 2-chloro-5-(chloromethyl)quinolin-8-ol hydrochloride(11)
2-Chloroquinolin-8-ol(3.42 g,19.04 mmol)was dissolved in 12N concentrated HCl(10 mL).37%Formal aldehyde solution(8 mL)and ZnCl2(1.10 g)were added.The reaction mixture was stirred at 40°C overnight.The precipitate was f i ltered and washed with copious acetone and dried under vacuum to give a yellow solid product,3.25 g,74.9%yield.
2-chloro-5-(((4-isopropylbenzyl)oxy)methyl)quinolin-8-ol(12)
2-Chloro-5-(chloromethyl)quinolin-8-ol hydrochloride(0.55 g,2.08 mmol)was suspended in 2 mL of 4-isopropylbenzyl alcohol.Thereaction mixturewasstirred and heated to 90°Cunder argon for 3 hours.Theresulted solution was stirred in 10 mL of saturated NaHCO3solution,extracted with ethyl acetate and washed with water.The extract was dried over anhydrous MgSO4followed by f i ltration and concentration under reduced pressure.The residue was puri f i ed by silica gel column chromatography(eluting with CH2Cl2)to giveacolorless oil product,0.70 g,98.5%yield.1HNMR(400 MHz,CDCl3)δ8.42(d,J=8.8 Hz,1H),7.77(s,1H),7.41(d,J=8.8 Hz,2H),7.26(s,1H),7.20(d,J=8.8 Hz,2H),7.13(d,J=8.0 Hz,1H),4.83(s,2H),4.49(s,2H),2.94-2.87(m,1H),1.24(d,J=6.8 Hz,6H).
Preparation of 2-chloro-5-(((4-isopropylbenzyl)oxy)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol(13)
2-chloro-5-(((4-isopropylbenzyl)oxy)methyl)quinolin-8-ol(0.50 g,1.46 mmol),paraformaldehyde(44 mg,1.46 mmol)and pyrrolidine(0.10 g,1.46 mmol)were mixed together in 10 mL of ethanol.The reaction mixture was stirred and heated to re f l ux for 4 hours under argon.Then,the volatile was removed under reduced pressure.The residue was puri f i ed by silica gel column chromatography(eluting with CH2Cl2/MeOH=9/1 v/v)to give a yellow solid product,0.56 g,90.3%yield.1HNMR(400 MHz,CDCl3)δ8.29(d,J=8.4 Hz,1H),7.35(d,J=8.4 Hz,1H),7.25(d,J=8.4 Hz,2H),7.21(d,J=8.4 Hz,2H),7.20(s,1H),4.80(s,2H),4.53(s,2H),3.99(s,2H),2.94-2.88(m,1H),2.75-2.71(m,4H),1.90-1.87(m,4H),1.25(d,J=6.8 Hz,6H).HRMS(ESI):m/z calculated for C25H29ClN2O2+H+[M+H]+:425.1996;Found:425.1997.
Preparation of 5-(((4-Isopropylbenzyl)oxy)methyl)-2-(4-methylpiperazin-1-yl)quinolin-8-ol(14)
2-Chloro-5-(((4-isopropylbenzyl)oxy)methyl)quinolin-8-ol(0.20 g,0.58 mmol)and(0.59 g,5.85 mmol)were mixed together in 5 mL of pyridine.The reaction mixture was stirred and heated to re f l ux for 8 hours under argon.Then,the volatile was removed under reduced pressure.Theresidue waspuri f i ed by silicagel column chromatography(eluting with CH2Cl2/MeOH=9/1 v/v)to give a yellow solid product,0.21 g,87.5%yield..1HNMR(400 MHz,CDCl3)δ8.21(d,J=9.2 Hz,1H),8.12(s,1H),7.13(d,J=8.4Hz,2H),7.08(d,J=8.4 Hz,2H),7.06(s,1H),6.97(d,J=9.2 Hz,1H),4.69(s,2H),4.36(s,2H),4.74(t,J=4.8 Hz,4H),2.94-2.87(m,1H),2.56(t,J=4.8 Hz,4H),2.37(s,3H),2.07-2.05(m,4H),1.20(d,J=6.8 Hz,6H).HRMS(ESI):m/z calculated for C25H31N3O2+H+[M+H]+:406.2495;Found:406.2485.
Preparation of 5-(((4-isopropylbenzyl)oxy)methyl)-2-(isopropylthio)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol(15)
Isopropyl thiol(0.21 g,2.94 mmol)was dissolved in anhydrous THF(40 mL).NaH(60%wt in mineral oil,0.14 g,3.54 mmol)was added in portions at 0°C under argon.After stirred at room temperature for 2 hours,2-Chloro-5-(((4-isopropylbenzyl)oxy)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol(13)(0.50 g,1.18 mmol)was added.The resulted mixture was stirred at room temperaturefor 6 hours.Then,thereaction wasquenched by addition of 50 mLof water.Themixturewasextracted with CH2Cl2(3×50 mL).The extracts was dried over anhydrous MgSO4,f i ltered and concentrated to dryness.The residue was puri f i ed by silica gel column chromatography(eluting with CH2Cl2/MeOH=9/1 v/v)to give a yellow solid product,0.46 mg,83.9%yield.1HNMR(400 MHz,CDCl3)δ8.15(d,J=8.8 Hz,1H),7.29(s,1H),7.23(d,J=8.8 Hz,2H),7.20(d,J=8.8 Hz,2H),7.19(d,J=8.0 Hz,2H),4.80(s,2H),4.50(s,2H),4.26-4.19(m,1H),4.00(s,2H),2.94-2.85(m,1H),2.78-2.76(m,4H),1.90-1.87(m,4H),1.49(d,J=6.4 Hz,6H),1.24(d,J=6.8 Hz,6H).HRMS (ESI):m/z calculated for C28H36N2O2S+H+[M+H]+:465.2576;Found:465.2580.
Preparation of 5-(((4-isopropylbenzyl)oxy)methyl)-2-(4-methylpiperazin-1-yl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol(16)
2-Chloro-5-(((4-isopropylbenzyl)oxy)methyl)-7-(pyridine-1-ylmethyl)quinolin-8-ol(13)(0.26 g,0.76 mmol)and 4-methylpiperazine(0.76 g,7.62 mmol)were mixed together in 5 mL of pyridine.The reaction mixture was stirred and heated to re f l ux for 2 hours under argon.Then,the volatile was removed under reduced pressure.The residuewas puri f i ed by silicagel column chromatography(eluting with CH2Cl2/MeOH=9/1 v/v)to give a yellow solid product,0.17 g,45.9%yield.1HNMR(400 MHz,CDCl3)δ8.15(d,J=9.2 Hz,1H),7.25(d,J=8.0 Hz,2H),7.20(d,J=8.0 Hz,2H),7.03(s,1H),6.74(d,J=9.2 Hz,1H),4.77(s,2H),4.47(s,2H),3.83(s,2H),3.60(t,J=6.4 Hz,4H),2.94-2.87(m,1H),2.75-2.72(m,4H),2.67-2.64(m,4H),2.38(s,3H),2.07-2.05(m,4H),1.24(d,J=6.8Hz,6H).HRMS(ESI):m/z calculated for C30H40N4O2+H+[M+H]+:489.3230;Found:489.3236.
Preparation of 7((1H-imidazol-1-yl)methyl-2-chloro-5-(((4-isopropylbenzyl)oxy)methyl)quinolin-8-ol(17)
2-Chloro-5-(((4-isopropylbenzyl)oxy)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol(13)(0.12 g,0.28 mmol)and imidazole(0.19 g,2.82 mmol)were mixed together in anhydrouspyridine(5 mL)under argon.The reaction mixture was stirred and heated to re f l ux for 6 hours under argon.Then,the volatile was removed under reduced pressure.The residue was puri f i ed by silica gel column chromatography(eluting with CH2Cl2/MeOH=19/1 v/v)to give a yellow solid product,65 mg,54.6%yield.1HNMR(400 MHz,CDCl3)δ8.56(s,H),8.41(d,J=9.2 Hz,1H),7.48(d,J=8.8 Hz,1H),7.25-7.23(m,6H),5.51(s,2H),4.82(s,2H),2.96-2.89(m,1H),2.16(s,2H),1.26(d,J=7.2 Hz,6H).HRMS (ESI):m/z calculated for C24H24ClN3O2+H+[M+H]+:422.1635;Found:422.1648.
Procedure for the preparation of compound 22
2-Hydroxyquinolin-8-yl acetate(18)
Quinoline-2,8-diol(5.00 g,31.00 mmol)was dissolved in anhydrous THF(30 mL)at room temperature under argon.Acetic anhydride(10 mL)wasadded via a syringe.The reaction solution was stirred and heated to re f l ux overnight.Then,the volatile was removed under reduced pressure.The residue was puri f i ed by silica gel column chromatography(eluting with CH2Cl2/MeOH=19/1 v/v)to give a white solid product,5.80 g,92.1%yield.
2-Bromoquinolin-8-yl acetate(19)
2-Hydroxyquinolin-8-yl acetate(2.50 g,12.30 mmol)was dissolved in chloroform(10 mL).POBr3(8.82 g,30.75 mmol)was added at room temperature.The reaction mixture was stirred and heated to re f l ux for 6 hours under argon.The reaction solution was cooled to roomtemperatureand poured into icewater followed by extraction with CH2Cl2,dried over anhydrous MgSO4/K2CO3,f i ltration and concentrated under reduced pressure.The residue was puri f i ed by silica gel column chromatography(eluting with CH2Cl2)to give a white solid product,2.80 g,86.0%yield.1HNMR(400 MHz,CDCl3)δ8.97(d,J=8.4 Hz,1H),7.69(dd,J=8.4,1.2 Hz,1H),7.57-7.52(m,2H),7.46(dd,J=8.4,1.2 Hz,1H),2.50(s,3H).
2-Bromo-5-(chloromethyl)quinolin-8-ol hydrochloride(20)
2-Bromoquinolin-8-yl acetate(1.20 g,4.51 mmol)was suspended in concentrated HCl(10 mL)at room temperature.ZnCl2(0.40 g)was added.The reaction mixturewasstirred and heated to 40°Cfor 8 hours.The yellow precipitate was separated,washed with acetone and dried under vacuum to givea yellow solid product,0.92 g,66.0%yield.1HNMR(400 MHz,DMSO-d6)δ 8.46(d,J=7.2 Hz,1H),7.79(d,J=8.8 Hz,1H),7.61(d,J=8.0 Hz,1H),7.11(d,J=8.0 Hz,1H),5.19(s,2H).
2-Bromo-5-(((4-isopropylbenzyl)oxy)methyl)quinolin-8-ol(21)
2-Bromo-5-(chloromethyl)quinolin-8-ol hydrochloride(0.50 g,1.62 mmol)was suspended in 5 mL of 4-isopropylbenzyl alcohol.The reaction mixture was stirred and heated to 90°Cunder argon for 4 hours.The resulted solution was stirred in 10 mL of saturated NaHCO3solution,extracted with ethyl acetate and washed with water.The extract was dried over anhydrous MgSO4followed by f i ltration and concentration under reduced pressure.Theresiduewaspuri f i ed by silica gel column chromatography(eluting with CH2Cl2)to give a colorless oil product,0.58 g,92.7%yield.1HNMR(400 MHz,CDCl3)δ8.41(d,J=8.8 Hz,1H),7.77(s,1H),7.41(d,J=8.8 Hz,2H),7.24(d,J=8.8 Hz,1H),7.20(d,J=8.8 Hz,2H),4.84(s,2H),4.40(s,2H),2.94-2.87(m,1H),1.24(d,J=7.2 Hz,6H).
2-Bromo-5-(((4-isopropylbenzyl)oxy)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol(22)
2-Bromo-5-(((4-isopropylbenzyl)oxy)methyl)quinolin-8-ol(0.84 g,2.17 mmol),paraformaldehyde(65 mg,2.17 mmol)and pyrrolidine(0.16 g,2.17 mmol)were mixed together in 10 mL of ethanol.The reaction mixture was stirred and heated to re f l ux for 4 hours under argon.Then,the volatile was removed under reduced pressure.The residue was puri f i ed by silica gel column chromatography(eluting with CH2Cl2/MeOH=9/1 v/v)to give a yellow solid product,0.74 g,72.6%yield.1HNMR(400 MHz,CDCl3)δ8.30(d,J=8.8 Hz,1H),7.36(d,J=8.8 Hz,1H),7.25(d,J=8.4 Hz,2H),7.21(d,J=8.4 Hz,2H),7.23(s,1H),4.80(s,2H),4.53(s,2H),4.01(s,2H),2.94-2.87(m,1H),2.74-2.72(m,4H),1.91-1.88(m,4H),1.24(d,J=6.8Hz,6H).HRMS(ESI):m/z calculated for C25H29BrN2O2+H+[M+H]+:469.1491;Found:469.1490.
Synthesis of 4-((tert-butyldimethylsilyl)oxy)-1-naphthaldehyde(24)
To a solution of aldehyde 15(2 mmol)in DCM was added TBDMSCl(3 mmol)and imidazole(4 mmol).Themixturewasstirred overnight.Water wasadded and themixturewasextracted with DCM.Theorganic layer was dried over anhydrous sodium sulfate.The crude waspuri f i ed by f l ash chromatography to generate24.1H NMR(400 MHz,Chloroform-d)δ10.12(s,1H),9.21(dt,J=8.7,1.0 Hz,1H),8.18(ddd,J=8.5,1.5,0.7 Hz,1H),7.77(d,J=7.9 Hz,1H),7.59(ddd,J=8.5,6.9,1.5 Hz,1H),7.48(ddd,J=8.3,6.9,1.2 Hz,1H),6.85(d,J=7.9 Hz,1H),1.01(s,9H),0.27(s,6H).
Synthesis of(4-((ter t-butyldimethylsilyl)oxy)naphthalen-1-yl)methanol(25)
To the protected aldehyde 16(1 mmol)in methanol wasadded NaBH4(3 mmol).Themixturewasstirred at room temperature for 2 hours.The solvent was evaporated and extracted with ethyl acetate.The crude wassubject to f l ash chromatography to producealcohol 25.1H NMR(400 MHz,Chloroform-d)δ8.24(ddd,J=8.1,1.5,0.7 Hz,1H),8.16-8.03(m,1H),7.52(dddd,J=22.0,8.1,6.8,1.4 Hz,2H),7.34(d,J=7.6 Hz,1H),6.81(d,J=7.7 Hz,1H),5.06(s,2H),1.10(s,9H),0.29(s,6H).
Synthesis of((4-((benzyloxy)methyl)naphthalen-1-yl)oxy)(tert-butyl)dimethylsilane(26)
To a solution of alcohol 25 in anhydrous THF(10ml)was added sodium hydride(60%dispersion in mineral oil)at 0°C.The suspension was stirred at 0°C for 30 minutes.Benzyl bromide was added to the suspension.The mixture was stirred at r.t for 3 hours.Water was added to the suspension,the mixture became homogeneous.Themixturewasextracted by ethyl acetateand washed with brine,dried over anhydroussodiumsulfate and concentrated to get the crude.The crude compound was puri f i ed by f l ash chromatography(ethyl acetate:hexane1:3).1HNMR(400 MHz,Chloroform-d)δ8.31-8.24(m,1H),7.92-7.82(m,1H),7.45-7.34(m,3H),7.34-7.27(m,3H),7.26-7.19(m,2H),6.73(d,J=7.9 Hz,1H),5.13(s,2H),5.00(s,2H),0.83(s,9H),0.00(s,6H).
Synthesis of 4-((benzyloxy)methyl)naphthalen-1-ol(27)
To a solution of alcohol 18 in THFwasadded TBAF(1.5 eq)and stirred for 1 hour.The mixture was extracted by ethyl acetate.The crude was subject to f l ash chromatography to produce phenol 27.1H NMR(400 MHz,DMSO-d6)δ10.21(s,1H),8.18(ddd,J=8.2,1.6,0.7 Hz,1H),8.02(ddd,J=8.4,1.3,0.7 Hz,1H),7.58-7.38(m,3H),7.36-7.25(m,5H),6.82(d,J=7.6 Hz,1H),4.84(s,2H),4.54(s,2H).
Synthesis of 4-((benzyloxy)methyl)-2-(pyrrolidin-1-ylmethyl)naphthalen-1-ol(28)
An equimolar mixture of the substrates 27,paraformaldehyde,and the pyrrolidine in anhydrous ethanol(30 mL)was re f l uxed for 4 hours under argon.After cooling,the solvent was evaporated under reduced pressure.The crude compound was puri f i ed by f l ash chromatography(Dichloromethane:methanol 20:1).1H NMR(400 MHz,Chloroform-d)δ8.31-8.23(m,1H),8.05-7.94(m,1H),7.57-7.44(m,2H),7.42-7.26(m,5H),7.10(s,1H),4.88(s,2H),4.61(s,2H),3.99(s,2H),2.89-2.52(m,4H),2.00-1.77(m,4H).Exact mass for C23H25NO2:347.1885;HRMS:[M+H]+:348.1959
Synthesis of 1-(benzyloxy)-4-((benzyloxy)methyl)benzene(30)
To a solution of alcohol 29(3 mmol)in anhydrous THF(30ml)wasadded sodiumhydride(60%dispersion in mineral oil,6 mmol)at 0°C.The suspension was stirred at 0°Cfor 30 minutes.Benzyl bromide(4 mmol)wasadded to thesuspension.Themixturewasstirred at r.t for 3 hours.Water was added to the suspension,the mixture became homogeneous.The mixture was extracted by ethyl acetate and washed with brine,dried over anhydrous sodium sulfate and concentrated to get the crude.The crude compound was puri f i ed by f l ash chromatography(ethyl acetate:hexane 1:3).1H NMR(400 MHz,Chloroform-d)δ7.52-7.18(m,12H),7.03-6.83(m,2H),5.07(s,2H),4.53(s,2H),4.49(s,2H).
Synthesis of 4-((benzyloxy)methyl)phenol(31)
To a solution of intermediate 12(1.0 equiv.)and NiCl2.6H2O(1.5 equiv.)in methanol(5 mL)at 0°C,NaBH4(3.0 equiv.)was added and stirred until complete consumption of the starting material.The progress of the reaction was monitored by TLC.After completion,the reaction mixture was quenched with methanol and stirred for another 20 minutes.Itwasthen f i ltered through celite pad and the f i ltrate was concentrated under reduced pressure.The crude product,thus obtained,was puri f i ed by column chromatography on activated silicagel using ethyl acetate-hexane mixtureaseluent to afford thepurephenol 13.1HNMR(400 MHz,Chloroform-d)δ7.39-7.33(m,4H),7.32-7.27(m,1H),7.26-7.21(m,2H),6.82-6.75(m,2H),4.54(s,2H),4.47(s,2H).
Synthesisfor 4-((benzyloxy)methyl)-2-(pyrrolidin-1-ylmethyl)phenol(32)
An equimolar mixture of the substrates 13,paraformaldehyde,and the pyrrolidine in anhydrous ethanol(30 mL)was re f l uxed for 4 hours under argon.After cooling,the solvent was evaporated under reduced pressure.The crude compound was puri f i ed by f l ash chromatography(Dichloromethane:methanol 20:1).1H NMR(400 MHz,Chloroform-d)δ7.39-7.33(m,4H),7.31-7.27(m,1H),7.13(dd,J=8.2,2.2 Hz,1H),6.99(d,J=2.2 Hz,1H),6.79(d,J=8.2 Hz,1H),4.54(s,2H),4.43(s,2H),3.82(s,2H),2.71-2.53(m,4H),1.91-1.80(m,4H).Exact mass for C19H23NO2:297.1729;HRMS:[M+H]+:298.1812
Synthesis of 5-(((4-isopropylbenzyl)oxy)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-yl tri f l uoromethanesulfonate(33)
To asolution of MX106 in anhydrous THFwasadded sodium hydrideat0°C.Themixturewasadded at0°Cfor 30 minutes.N-Phenyl-bis(tri f l uoromethanesulfonimide)wasadded.Themixturewasre f l uxed for 4hr.Themixture was extracted by ethyl acetate.The crude was subject to f l ash chromatography to generate 33.1H NMR(400 MHz,Chloroform-d)δ9.00(dd,J=4.2,1.6 Hz,1H),8.47(dd,J=8.6,1.6 Hz,1H),7.84(s,1H),7.51(dd,J=8.5,4.2 Hz,1H),7.29(d,J=8.2 Hz,2H),7.24(d,J=8.2 Hz,2H),4.95(s,2H),4.59(s,2H),3.95(s,2H),2.93(p,J=6.9 Hz,1H),2.74-2.44(m,4H),1.96-1.71(m,4H),1.26(d,J=6.9 Hz,6H).Exactmassfor C26H29F3N2O4S:522.1800;HRMS:[M+H]+:523.1884
Synthesis of 5-(((4-isopropylbenzyl)oxy)methyl)-7-(pyrrolidin-1-ylmethyl)quinoline(34)
To asolution of compound 33 in methanol wasadded Pd/c(10%,0.05eq),Mg(0.1 eq)and ammoniumacetate(2 eq).The mixture wasstirred at room temperate for 5 hours.Themixturewas f i ltered by celite.Thecrudewas puri f i ed to generatecompound 25.1H NMR(400 MHz,Chloroform-d)δ8.93-8.77(m,1H),8.59-8.45(m,1H),8.07(s,1H),7.82(d,J=1.7 Hz,1H),7.46(dd,J=8.6,4.3 Hz,1H),7.20(d,J=7.5 Hz,3H),7.13(d,J=8.2 Hz,2H),4.91(s,2H),4.52(s,2H),4.41(s,2H),3.57(s,2H),3.06(s,2H),2.82(p,J=6.9 Hz,1H),2.08(s,5H),1.16(d,J=6.9 Hz,6H).Exact mass for C25H30N2O:374.2358;HRMS:[M+H]+:375.2439
Synthesis for 5-(((4-isopropylbenzyl)oxy)methyl)-7-(pyrrolidin-1-ylmethyl)-1,2,3,4-tetrahydroquinoline(35)
To asolution of compound 33 in methanol wasadded Pd/C(10%,0.1eq),Mg(0.2 eq)and ammonium acetate(2 eq).The mixture was stirred at room temperate overnight.Themixturewas f i ltered by celite.Thecrude was puri f i ed to generate compound 35.1H NMR(400 MHz,Chloroform-d)δ9.95(s,1H),7.21(d,J=8.2 Hz,2H),7.17-7.13(m,2H),6.67(d,J=1.9 Hz,1H),6.52(d,J=1.9 Hz,1H),4.46(s,2H),4.37(s,2H),3.96(s,2H),3.56(s,3H),3.24-3.12(m,2H),2.94-2.74(m,2H),2.60(t,J=6.5 Hz,2H),2.11(d,J=8.5 Hz,2H),1.96(d,J=7.9 Hz,2H),1.84(td,J=6.4,4.6 Hz,2H),1.18(d,J=6.9 Hz,6H).Exact mass for C25H34N2O:378.2671;HRMS:[M+H]+:379.2750
General procedure for the synthesis of compounds(36a-b)
To a solution of alcohol(6 mmol)in anhydrous THF(30ml)was added sodium hydride(60%dispersion in mineral oil,0.72 g,18 mmol)at 0°C.The suspension wasstirred at0°Cfor 30 minutes.Salt2(1.15 g,5mmol)wasadded to thesuspension.Themixturewasstirred at r.t for 3 hours.Water was added to the suspension,the mixture became homogeneous.The mixture was extracted by ethyl acetate and washed with brine,dried over anhydrous sodium sulfate and concentrated to get the crude.The crude compound was puri f i ed by f l ash chromatography(ethyl acetate:hexane 1:3)
(R)-5-((1-(2,6-dichloro-3-f l uorophenyl)ethoxy)methyl)quinolin-8-ol(36a)
1H NMR(400 MHz,Chloroform-d)δ8.83(dd,J=4.3,1.6 Hz,1H),8.59(dd,J=8.6,1.6 Hz,1H),7.54(dd,J=8.5,4.3 Hz,1H),7.35(d,J=7.7 Hz,1H),7.28-7.25(m,1H),7.12(d,J=7.7 Hz,1H),7.05(ddd,J=8.9,8.0,4.6 Hz,1H),5.34(q,J=6.8 Hz,1H),4.85(d,J=11.8 Hz,1H),4.66(d,J=11.8 Hz,1H),1.60(d,J=6.8 Hz,3H).
(S)-5-((1-(2,6-dichloro-3-f l uorophenyl)ethoxy)methyl)quinolin-8-ol(36b)
1H NMR(400 MHz,Chloroform-d)δ8.78(dd,J=4.2,1.6 Hz,1H),8.52(dd,J=8.5,1.5 Hz,1H),7.49(dd,J=8.5,4.2 Hz,1H),7.33-7.27(m,1H),7.25-7.22(m,1H),7.07-7.04(m,1H),7.03-6.99(m,1H),5.58(s,1H),5.31(q,J=6.8 Hz,1H),4.82(dd,J=11.7,0.6 Hz,1H),4.62(d,J=11.8 Hz,1H),1.65(d,J=6.9 Hz,3H).
General procedure for the synthesis of compounds(37a-b)
An equimolar mixture of the substrates 36,paraformaldehyde,and the pyrrolidine in anhydrous ethanol(30 mL)was re f l uxed for 4 hours under argon.After cooling,the solvent was evaporated under reduced pressure.The crude compound was puri f i ed by f l ash chromatography(Dichloromethane:methanol 20:1).
(R)-5-((1-(2,6-dichloro-3-f l uorophenyl)ethoxy)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol(37a)
1H NMR(400 MHz,Chloroform-d)δ8.79(dd,J=4.2,1.7 Hz,1H),8.38(dd,J=8.5,1.7 Hz,1H),7.35(dd,J=8.5,4.2 Hz,1H),7.13(dd,J=8.9,4.9 Hz,1H),7.05(s,1H),6.92(dd,J=8.9,8.0 Hz,1H),5.24-5.16(m,2H),4.73(d,J=11.8 Hz,1H),4.56(d,J=11.8 Hz,1H),3.92(d,J=1.6 Hz,2H),2.76-2.55(m,4H),1.88-1.73(m,4H),1.50(d,J=6.8 Hz,3H).Exact massfor C23H23Cl2FN2O2:448.1121;HRMS:[M+H]+:449.1191
(S)-5-((1-(2,6-dichloro-3-f l uorophenyl)ethoxy)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol(37b)
1H NMR(400 MHz,Chloroform-d)δ8.87(dd,J=4.1,1.6 Hz,1H),8.45(dd,J=8.5,1.7 Hz,1H),7.41(dd,J=8.5,4.2 Hz,1H),7.20(dd,J=8.8,4.9 Hz,1H),7.09(s,1H),6.99(dd,J=8.9,8.0 Hz,1H),5.35-5.22(m,2H),4.80(d,J=11.8 Hz,1H),4.62(d,J=11.8 Hz,1H),3.97(s,2H),2.81-2.59(m,4H),1.95-1.77(m,4H),1.57(d,J=6.8 Hz,3H).Exact mass for C23H23Cl2FN2O2:448.1121;HRMS:[M+H]+:449.1205
Synthesis of 5-(((4-tri f l uoromethylbenzyl)amino)methyl)quinolin-8-ol(38)
5-(Chloromethyl)quinolin-8-ol hydrochloride(0.50 g,2.17 mmol)and 4-tri f l uorobenzylamine(1.14 g,6.52 mmol)were mixed together in 40 mL of ethyl acetate/DMF(1/3).Thereaction mixturewasstirred and heated to 60°C for 24 hours.The reaction was quenched by addition of 50 mL of saturated sodium bicarbonate solution and extracted with CH2Cl2(3×50 mL).The extracts was dried over anhydrous MgSO4followed by f i ltration and concentration under reduced pressure.The residue was puri f i ed by silica gel column chromatography(eluting with CH2Cl2/MeOH 19/1 v/v)to give a gray solid,0.26 g,36.1%yield.1HNMR(400 MHz,CDCl3)δ8.81(d,J=3.2 Hz,1H),8.52(d,J=8.4 Hz,1H),7.61(d,J=8.0 Hz,2H),7.49(d,J=8.0 Hz,2H),7.41(d,J=8.0Hz,1H),7.11(d,J=8.0 Hz,1H),4.16(s,2H),3.95(s,2H).
Synthesis of 2-bromo-5-(((4-isopropylbenzyl)oxy)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol.(39)
5-(((4-tri f l uoromethylbenzyl)amino)methyl)quinolin-8-ol(0.16 g,0.48 mmol),para-formaldehyde(14 mg,0.48 mmol)and pyrrolidine(34 mg,0.48 mmol)were mixed together in 10 mL of ethanol.The reaction mixture was stirred and heated to re f l ux for 4 hours under argon.Then,the volatile was removed under reduced pressure.The residue was puri f i ed by silica gel column chromatography(eluting with CH2Cl2/MeOH=9/1 v/v)to give a yellow solid product,0.14 g,70.4%yield.1HNMR(400 MHz,CDCl3)δ8.79(d,J=3.6 Hz,1H),8.01(dd,J=8.4,1.6 Hz,1H),7.50(d,J=7.2 Hz,1H),7.31(d,J=8.4 Hz,2H),7.27(d,J=8.4 Hz,2H),7.15(s,1H),3.96(s,2H),3.57(s,2H),3.28(s,2H),2.68-2.65(m,4H),1.82-1.80(m,4H).HRMS(ESI):m/z calculated for C23H24F3N3O+H+[M+H]+:416.1950;Found:416.1965.
Cell culture and in vitro antiproliferative assays in human melanoma cells
Human melanoma A375 and M14 cell lines were purchased from ATCC(American Type Culture Collection,Manassas,VA,USA),and cultured in DMEM media(Mediatech,Inc.,Manassas,VA)at 37°C in a humidi f i ed atmospherecontaining 5%CO2.Theculture mediaweresupplemented with 10%fetal bovineserum(Atlanta Biologicals,Lawrenceville,GA)and 1%antibiotic-antimycotic mixture(Sigma-Aldrich,St.Louis,MO).Compounds were dissolved in dimethylsulfoxide(DMSO;Sigma-Aldrich)to make a stock solution of 10 mmol/L.Compound solutions were freshly prepared by diluting stocks with cell culture medium before use(f i nal solution contained less than 0.5%DMSO).Fivethousand cellsin logarithm growing phasewereseeded overnight into each well of a96-well plate.Then thecellswerecontinuously incubated for 48 hours with sequential diluted compound solution(3 nmol/L to 100μmol/L,100μL per well)in cell culture medium.The cell viability was determined in MTS assay and IC50wascalculated(n=4),following similar procedures as described previously[25-28].
Cell culture and cytotoxicity assays in human epidermoid carcinoma and colorectal cancer cells
Dulbecco’smodi f i ed Eagle's Medium(DMEM),fetal bovine serum(FBS),penicillin/streptomycin and trypsin 0.25%were purchased from Hyclone(GE Healthcare Life Science,Pittsburgh,PA).Phosphate buffered saline(PBS)was purchased from Invitrogen GIBCO(Grand Island,NY).Dimethyl sulfoxide(DMSO)and 3-(4,5-dimethylthiazole-2-yl)-2,5-biphenyl tetrazolium bromide(MTT)were purchased from Sigma Chemical Co(St.Louis,MO).
The P-glycoprotein(P-gp)overexpressing KB-C2 cell line was established from a parental human epidermoid carcinoma cell line KB-3-1,by a stepwiseselection of KB-3-1in increasing concentrationsof colchicine up to 2μg/mL[29].SW620/Ad300,which is also a P-gp overexpressing drug resistant cell line,was established by stepwiseexposureof theparental human colon cancer cell line SW620 to increasing concentrations of doxorubicin up to 300 ng/mL[30].The SW620 and SW620/Ad300 cell lines were kindly provided by Dr.Susan E.Batesand Dr.Robert W.Robey(NCI,NIH,Bethesda,MD,USA).All the cell lines were grown in DMEM supplemented with 10%FBSand 100 unit/mL penicillin/streptomycin in a humidi f i ed incubator containing 5%CO2at 37°C.
The MTTcolorimetric assay wasused to measurethe sensitivity of the cells against the synthesized compounds.Theassay detects the formazan product formed from the reduction of MTT in active cells thus assesses the cell viability[31].Cellswere seeded in 96-well plates at 5,000 cells/well(KB-3-1 or KB-C2 cells)or at 7000 cells/well(SW620 or SW620/Ad300 cells)in 180μL completed medium and cultured overnight.Then various concentrations of the compounds(20μL)were added to the designated wells.After 72 hours continuous drug incubation,20μL of MTT reagent(4 mg/mL)was added to each well and the plates were incubated at 37°C for 4 hours.Subsequently,the medium was removed and 100µL of DMSO were added to dissolve the formazan crystals in each well.The absorbance was determined at 570 nm by the accuSkanTMGOUV/Vis Microplate Spectrophotometer(Fisher Sci.,Fair Lawn,NJ).The IC50values of each compound on each cell line were calculated from the survival curves to represent the cytotoxicity of the compounds.The fold of drug resistance was calculated by dividing the IC50of the P-gp overexpressing cellsby that of the parental cells.Two known P-gp substrates,YM155 and paclitaxel,were used as positive controls for P-gp overexpressing cell lines.On the other hand,cisplatin,which is not a substrate of P-gp,was used as negative control drug.
Western blotting
To determine the change of protein levelsof survivin and closely related IAPs,lysates of A375 or M14 melanomacellstreated by thecompound solution for 24 hours were used for Western blotting analysis.Primary rabbit antibodies against IAPproteins including survivin(#2808),XIAP(#2045),cIAP1(#7065),Livin(#5471),Cleaved PARP(#9185)and theloading control protein GAPDH(HRP Conjugate)(#3683)were purchased from Cell Signaling Technology,Inc.(Danvers,MA)and used according to manufacture instructions as reported previously.Protein lane intensities were quanti f i ed using ImageJ software(US National Institutes of Health,Bethesda,MD)and band intensities were normalized with vehicle control and GAPDH loading control.
Statistical analysis
All experimentswererepeated atleastthreetimesand the differences between the mean values of two groups were determined by using two-tailed Student's t-test in cell cytotoxicity assay.The difference between the survival curves of two groupsweredetermined by twoway ANOVA.Results were considered statistically signi f i cant when P<0.05.
Results
Chemistry
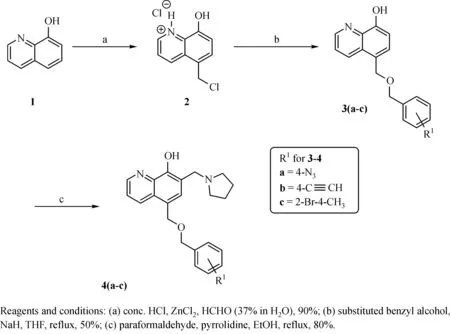
Fig.2 Synthesis of Compounds 4a-4c
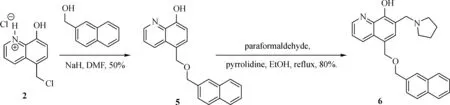
Fig.3 Synthesis of Compound 6
The general synthesis of C ring substituted UC112 analogs 4a-4c is shown in Fig.2.The procedure is similar to the one reported in our recent f i ndings[26].First,salt 2 was generated,which then reacted with different substituted benzyl alcohols to form ethers 3a-3c.Then ether 3a-3c were submitted to Mannich reaction with paraformaldehyde and pyrrolidine to form compounds 4a-4c.The synthesis of compound 6 applied similar method as compound 4a-4c,which is shown in Fig.3.Compound 8 which has morpholine ring on the C ring position was prepared as Fig.4 shown.First,salt 2 reacted with morpholine to form intermediate 7.Then intermediate 7 was converted to compound 8 through Mannich reaction.The synthesis of compounds 13-17 is outlined in Fig.5.Quinoline-2,8-diol f i rst reacted with thionyl chloride in DMF to generate intermediate 10,which was converted to intermediate 11 by reacting with formaldehyde and catalytic zinc chloride in concentrated hydrochloric acid.Then intermediate 11 was heated in 4-isopropylbenzyl alcohol to form ether 12.After ether 12 is obtained,it underwent two ways.In the f i rst way,ether 12 went through Mannich reaction to form compound 13.In the second way,the chloro group on A ring of ether 12 isreplaced with 4-methylpiperazineto generate compound 14.Compounds 15-17 are synthesized from compound 13.Compound 15 wasproduced through the reaction of Compound 13 with Isopropyl thiol.Compound 16 was obtained through the reaction of 4-methylpiperazine with Compound 13.Replacing the pyrrolidine ring on Compound 13 with imidazole ring generated compound 17.Compound 22,which has bromo on A ring,was prepared as Fig.6 shown.The hydroxyl group on quinoline-2,8-diol was protected by acetate group[32].Then,the bromo group was introduced to A ring to form intermediate 19,which was converted to intermediate 20 by reacting with formaldehyde and catalytic zinc chloride in concentrated hydrochloric acid.Intermediate 20 washeated in heated in 4-isopropylbenzyl alcohol to form ether 21.Ether 21 then underwent Mannich reaction to producecompound 22.Compound 28 which replacesthenitrogen on the A ring with Carbon was synthesized as Fig.7 shown.Aldehyde 23 was f i rst protected by TBDMS group.Then the protected aldehyde 24 was reduced to form alcohol 25,which reacted with benzyl bromide to form ether 26.De-protection of ether 26 with TBAF generated intermediate 27.Then intermediate 27 was converted to compound 28 via Mannich reaction.Compound 32 which has no A ring was prepared as Fig.8 shown.Alcohol 29 was f i rst reacted with benzyl bromide to form ether 30,which was converted to intermediate 31 by removing the benzyl group on phenol position using the method reported in literature[33].Intermediate 31 then underwent Mannich reaction to generate compound 32.The synthesis of compound 34 and 35 isshown Fig.9.Firsttri f l ategroup was introduced to MX106 to form compound 33.Removal of the OTf group produced compound 34.Reduction of compound 33 with Pd/Carbon generated compound 35.Compounds37a-b wereprepared as Fig.10 shown.First ethers 36a-b were synthesized.Then they underwent Mannich reaction to form compounds 37a-b.The amine linked analog,compound 39 was prepared as Fig.11 shown.Salt 2 f i rst reacted with amineto form intermediate 38,which was converted to compound 39 through Mannich reaction.
Biological Results
Effect of C ring modi f i cation on MX106
The in vitro activity for C ring modi f i ed MX106 analogs,compounds 4a-4c,6 and 8 in A375 and M14 melanoma cells is shown in Table 1.Replacing the isopropyl group with an azide group(4a)or an ethynyl group(4b)resulted in lower activity than MX106 in both cell lines(1.6µmol/L and 1.41µmol/L vs.0.73µmol/L in A375;1.4µmol/L,and 1.38µmol/L vs.0.72µmol/L in M14,for 4a,4b,and MX106,respectively).However,introducing two substitution groups(4c)to the C-ring or replacing the substituted phenyl ring with a naphthalene ring(6)maintain the activity compared with that of MX106 in both A375 cell line(0.75µmol/L and 0.82µmol/L vs.0.73µmol/L,for 4c and 6,respectively)and M 14 cell(0.75µmol/L and 0.62µmol/L vs.0.72µmol/L).These results suggest that a more lipophilic group at C ring position is slightly favorable for activity.Consistent with this,compound 8 which replaced the phenyl ring with less lipophilic ring morpholinecaused signi f i cantly decrease of activity(29.09µmol/L vs.0.73µmol/L in A375,16.65µmol/L vs.0.73µmol/L in M14).These results further con f i rmed our previous SAR studies on this ring[26].
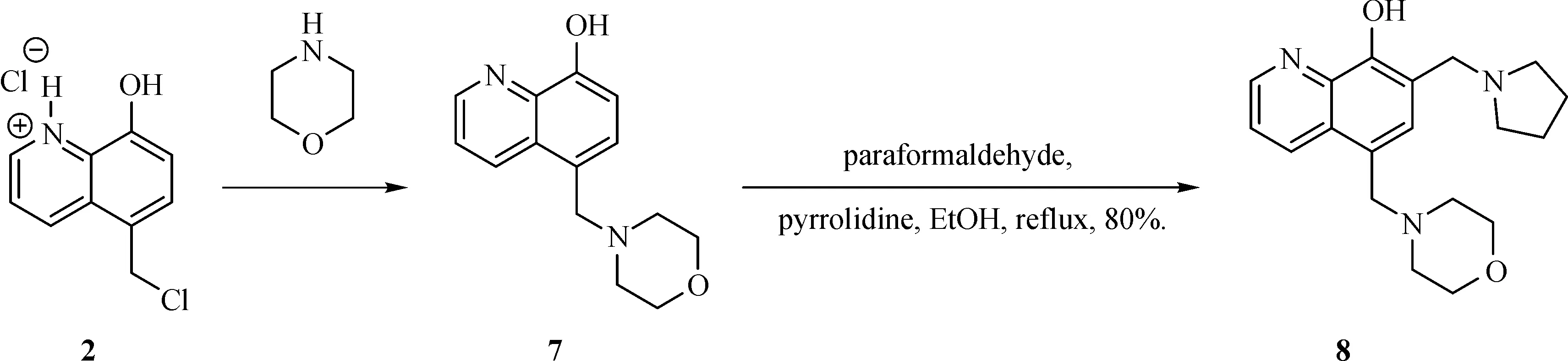
Fig.4 Synthesis of Compound 8
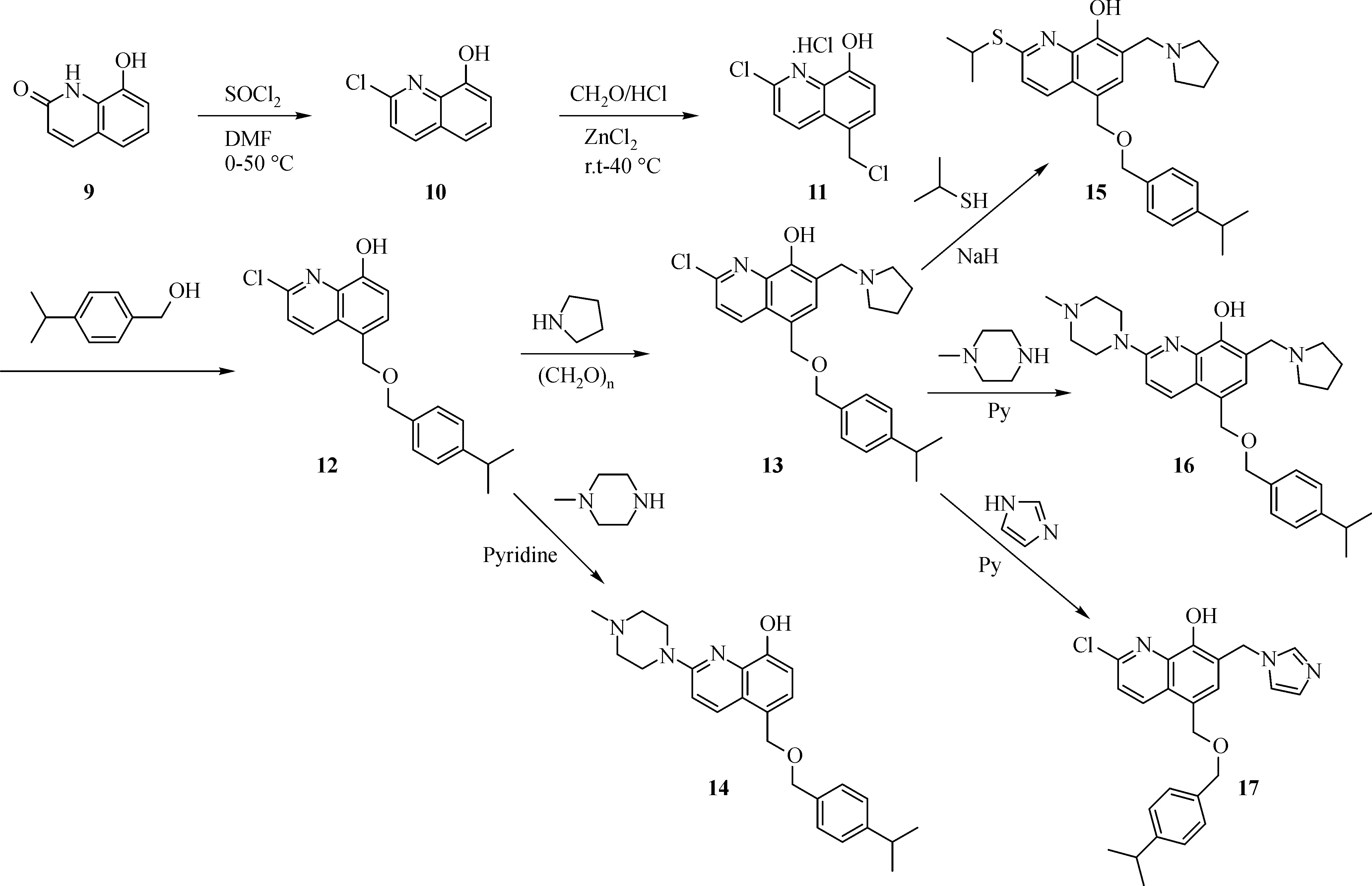
Fig.5 Synthesis of Compounds 13-17
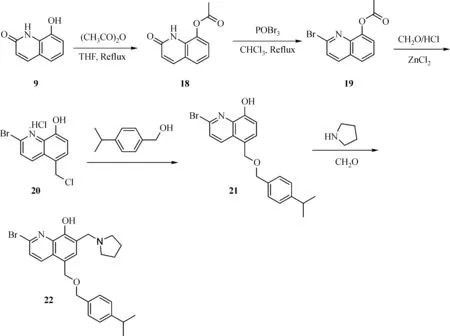
Fig.6 Synthesis of Compound 22
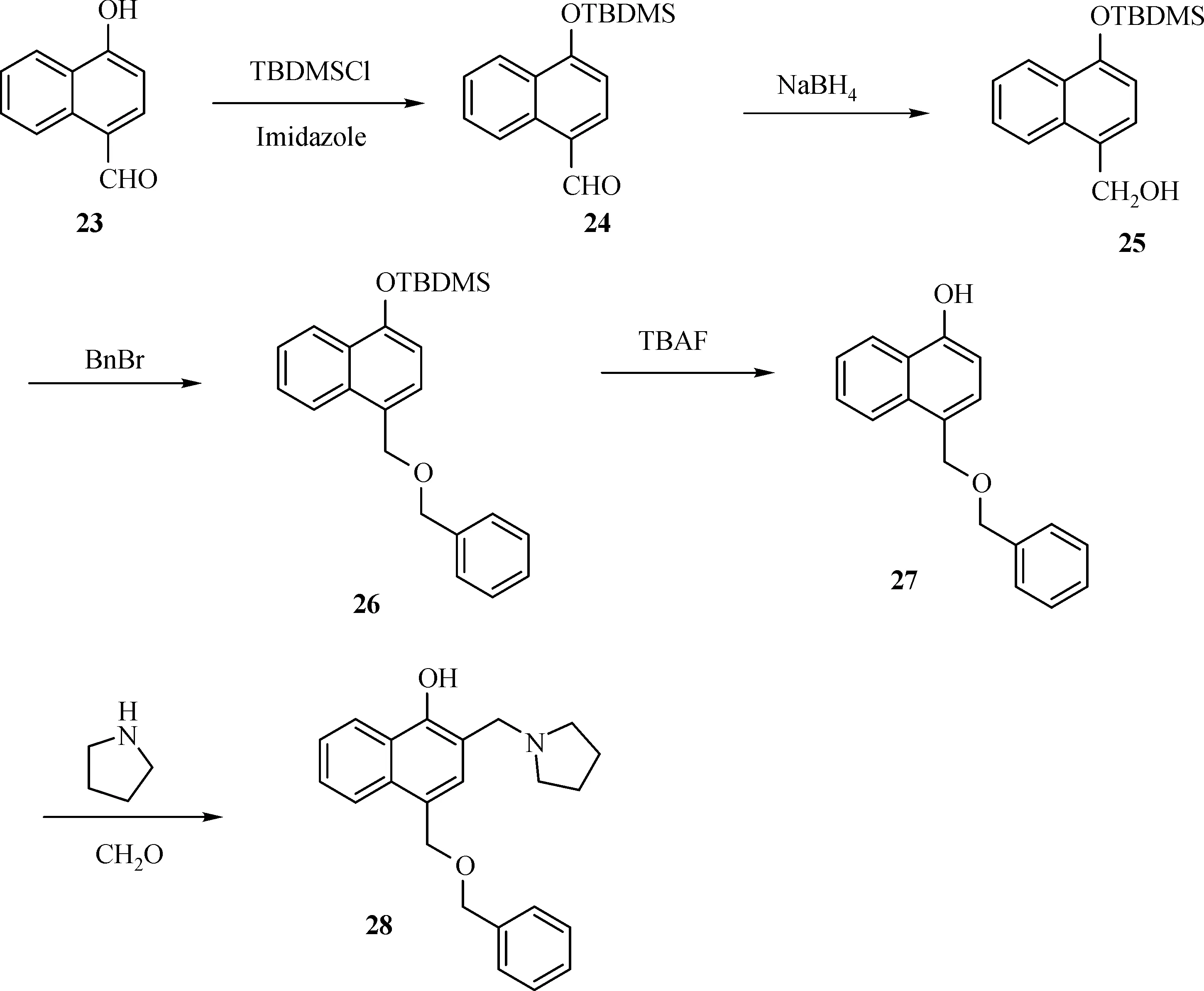
Fig.7 Synthesis of Compound 28
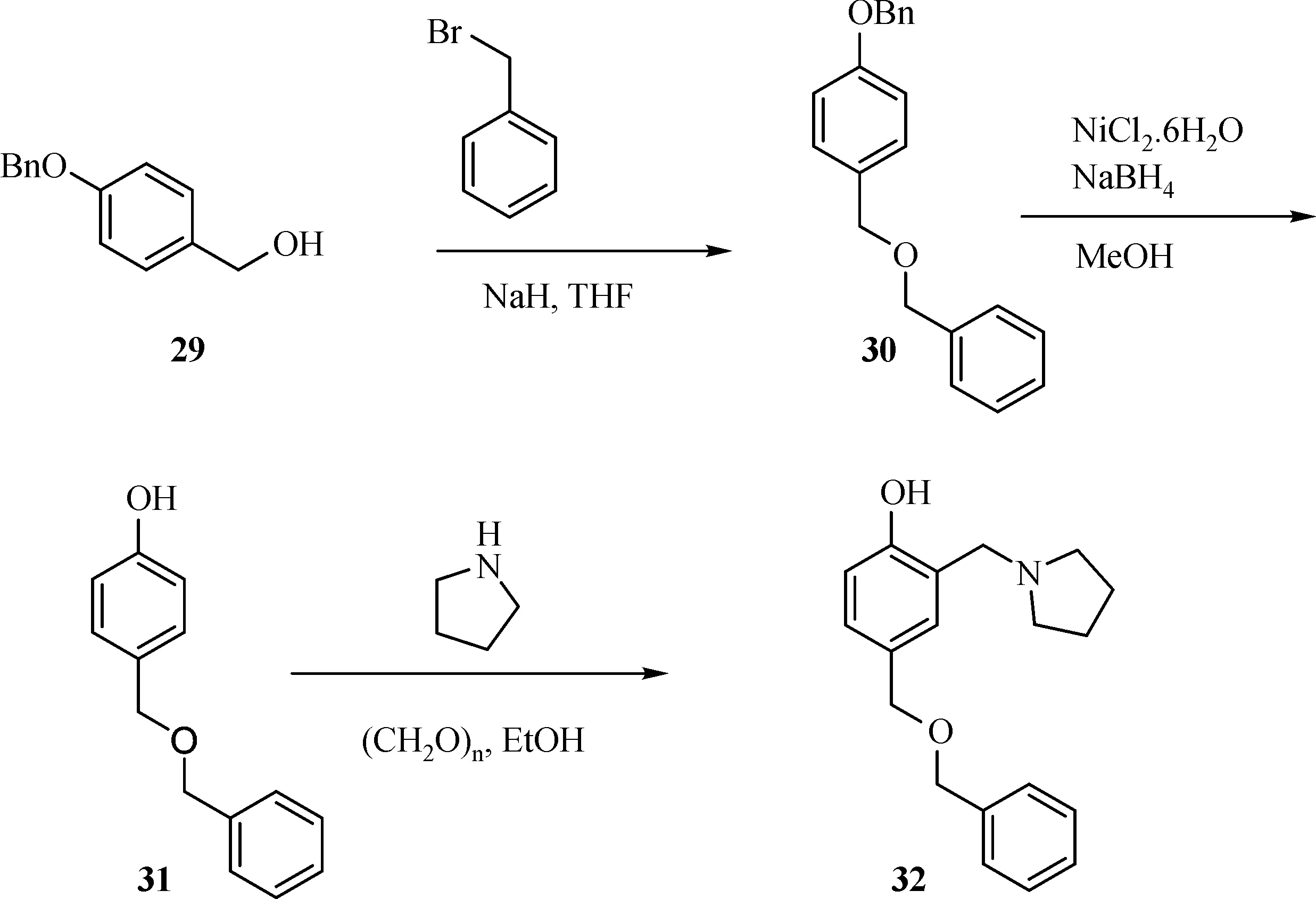
Fig.8 Synthesis of Compound 32
Effect of Aring modi f i cation on MX106
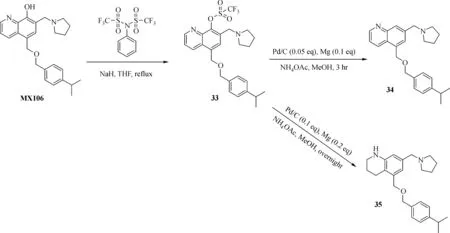
Fig.9 Synthesis of Compounds 33-35
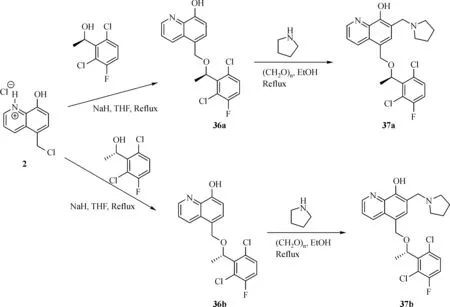
Fig.10 Synthesis of Compounds 37a-b
As shown in Table 1,all these modi f i cations resulted in signi f i cantly loss of activity.Introducing a chloro(13),a bromo(22),or an isopropyl thiol group(15)to the A ring of MX106 all decreasestheanti-proliferative activity compared with that of MX106.The addition of a bulky group as 4-methylpiperazine further decreased activity.Furthermore,replacing the nitrogen atom with acarbon atomonthe A ring(28)or completeremoval of thisring(32)caused total lossof activity.These in vitro results demonstrate that introduction of substitutions or removal of thispyridinering of MX106 isnot tolerable,and that an unsubstituted pyridine ring on the A ring positon is optimal for activity.
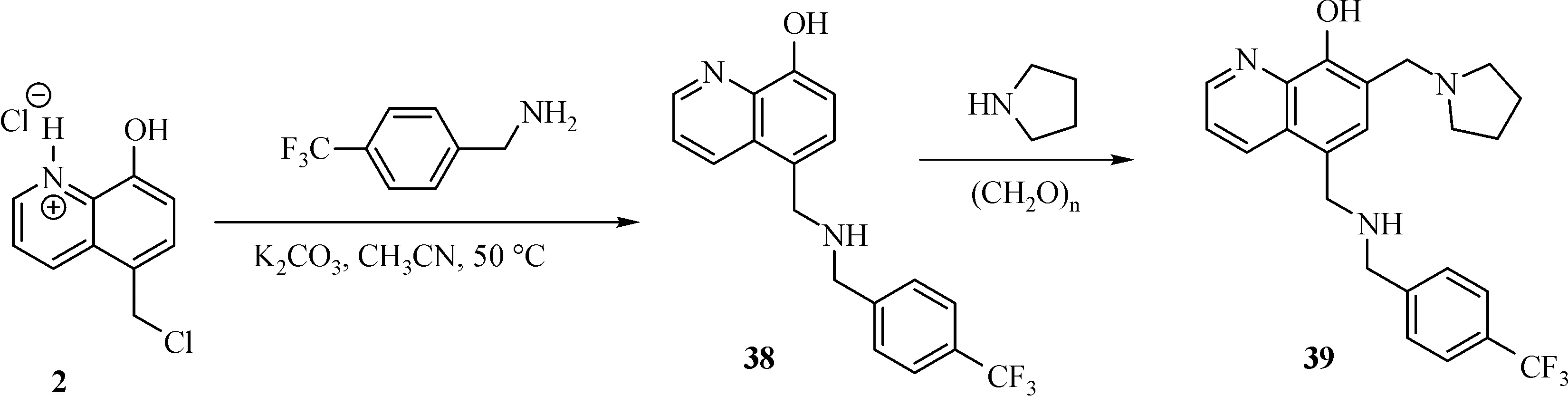
Fig.11 Synthesis of Compound 39
Effect of D ring modi f i cation on MX106
Some D ring modi f i cations were also performed based on the A ring modi f i ed analogs.The in vitro activity for the D ring modi f i ed analogs,compounds14 and 17 are also shown in Table 1.Compound 14 which removed D ring did not improve activity compared to MX106.Compound 17 which replaced pyrrolidine ring with imidazolering waslessactivethan MX106 aswell.This result suggests the pyrrolidine ring in this scaffold is needed for activity.
Effect of B ring modi f i cation on MX106
Since B ring on the UC112 scaffold haven't been modi f i ed before,we made several B ring modi f i ed analogs,compound 33-35.As shown in Table 1,Compound 33 which protects the hydroxyl group on the B ring with tri f l ate group was basically inactive,which means the free hydroxyl group is essential for activity.Compound 34,which removes the hydroxyl group on the B ring,was also inactive.This further con f i rmstherequirement of afreehydroxyl group on B ring for activity.Compound 35 which reduced the pyridine ring to a saturated ring improved activity compared with itsunsaturated form 34,but it isstill less active than MX106.
Effect of linkers between B ring and C ring of MX106
Finally,we modi f i ed the linker between B ring and C ring of MX106.Threecompounds37a,37b and 39 weremade as a result.As shown in Table 1.Compound 37a which introduced a methyl group to the linker was slightly moreactive than MX106 in A375(0.65µmol/L vs.0.73µmol/L in A375)and has equivalent activity in M14cell.Theadded methyl group makesthelinker more conformationally rigid,which may explain for the increase of activity.The stereo isomer 37b was slightly lessactivethan MX106(0.80µmol/L vs.0.73µmol/L in A375,0.83µmol/L vs.0.72µmol/L in M14),which suggests there might be a steric requirement for activity.Replacing the oxygen atom in MX106 with NH(compound 39)decreases activity(3.44µmol/L vs.0.73µmol/L in A375,3.80µmol/L vs.0.72µmol/L in M14).
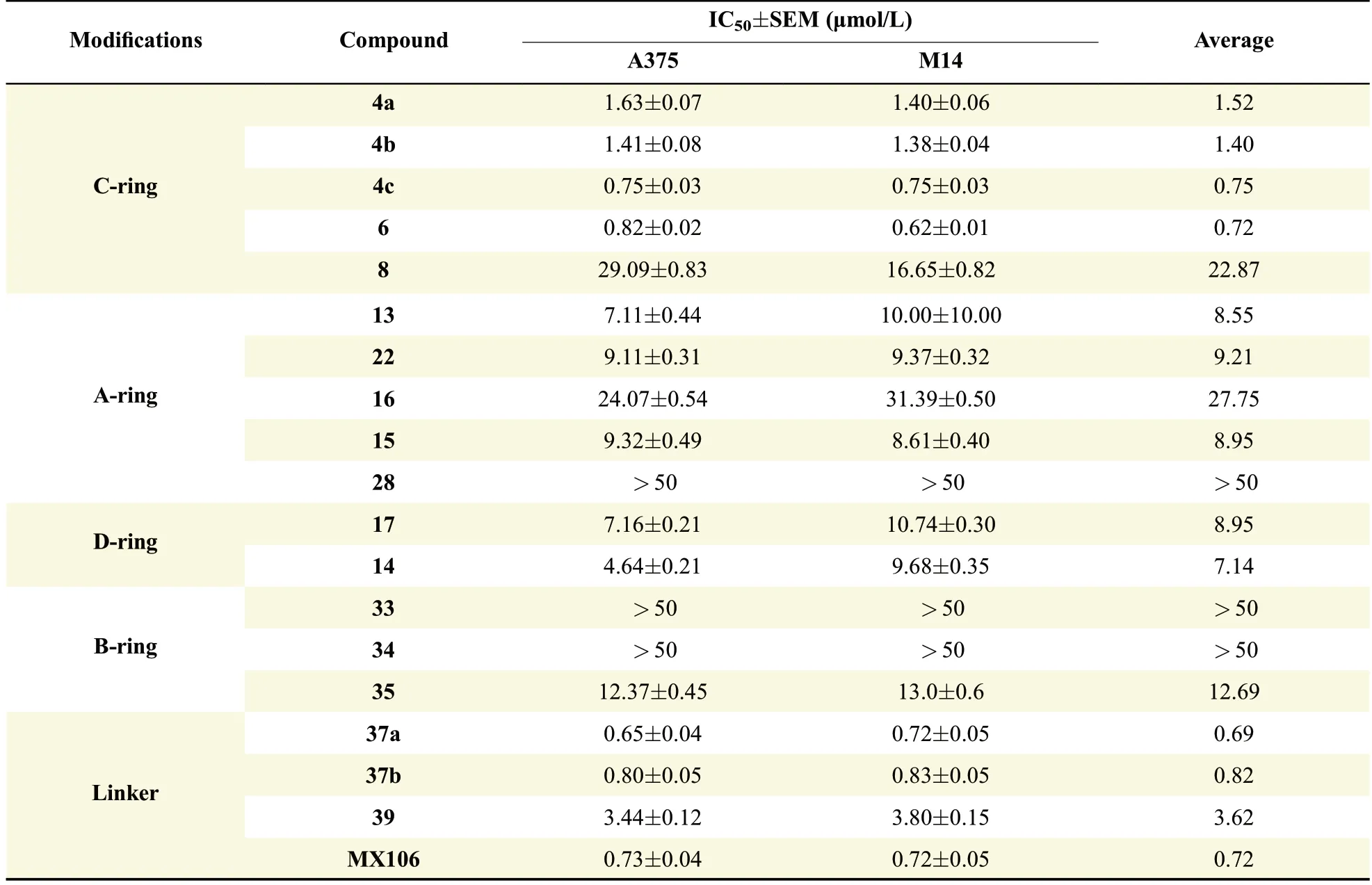
Table 1 Effects of structural modi f i cations for MX106 on antiproliferative activity.
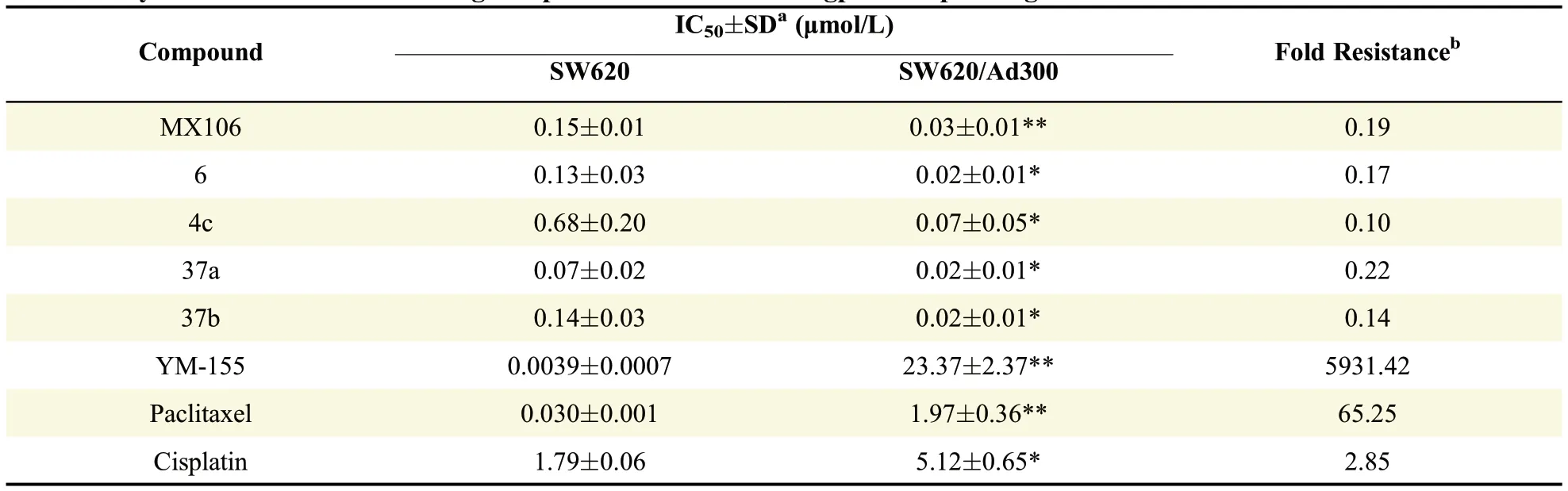
Table 2 Cytotoxic effects of f i ve analogs on parental SW620 and P-gp overexpressing SW620/Ad300 cell lines.
In general,this systematic modi f i cation on different parts of MX106 did not provide signi f i cant improvement to its activity,suggesting there are limited structural modi f i cations that can be tolerated in this scaffold to maintain the ef f i ciency for disrupting protein-protein interactions.
MDRevaluationresultsofMX106analogs
In order to evaluatewhether our new compoundscan overcome MDR,we selected four most potent compounds,4c,6,37a and 37b along with MX106,and submitted themto two pairsof cell linesfor in vitro antiproliferative activity test.One pair was P-gp overexpressed SW620/Ad300 cell line and its parental,drug-sensitivecell line SW620.Theother pair was P-gp overexpressed KB-C2 cell line and its parental,drugsensitive cell line KB-3-1.The IC50values of the f i ve compoundson theparental cancer cell linesand their Pgp overexpressing resistant sublines were summarized in Table 2 and Table 3.From Table 2,all MX106 analogs showed signi f i cantly higher potency in drug resistant cell line SW620/Ad300 than its parental cell line SW620.The f i ve analogs all had very small values of fold resistance.Compound 37a has a fold resistance as low as 0.07.Similarly,as shown in Table 3,MX106 analogsweremoreactivein drug resistant cell line KB-C2 than itsparental cell line KB-3-1.Although existing anticancer drug paclitaxel and survivin inhibitor YM155 were more potent than MX106 analogs in parental sensitive cell lines SW620 and KB-3-1,they essentially lost activities in resistant cell lines SW620/Ad300 and KB-C2.For P-gp substrates YM155 and paclitaxel,strong drug resistance was observed on SW620/Ad300 cell line(5931.42-and 65.25-fold,respectively)and on KB-C2 cell line(7549.38-and 529.42-fold,respectively).On the contrary,cisplatin,which is not a substrate of P-gp,showed similar sensitivity or low resistance(1.8-to 5.1-fold)in drug resistant cells compared to the parental cells.Similarly,the survival curves were made by plotting the survival rate of cell lines at various drug concentrations(Supplemental Fig.S1 and Supplemental Fig.S2).As shown in Fig.S1 and Fig.S2,P-gp substrates YM155 and paclitaxel made an obvious shift to right in survival curves of KB-C2 and SW620/Ad300 in comparison with those of the parental cell lines KB-3-1 and SW620,whereas cisplatin exhibited close survival curves between parental and resistant cell lines.In contrast,P-gp overexpressing cell lines treated with our analogs had the survival curves shifting to left compared to their parental cell lines,indicating that the analogs were able to not only overcome the MDR resulting from P-gp overexpression,but also kill the resistant cells at a lower drug concentration.
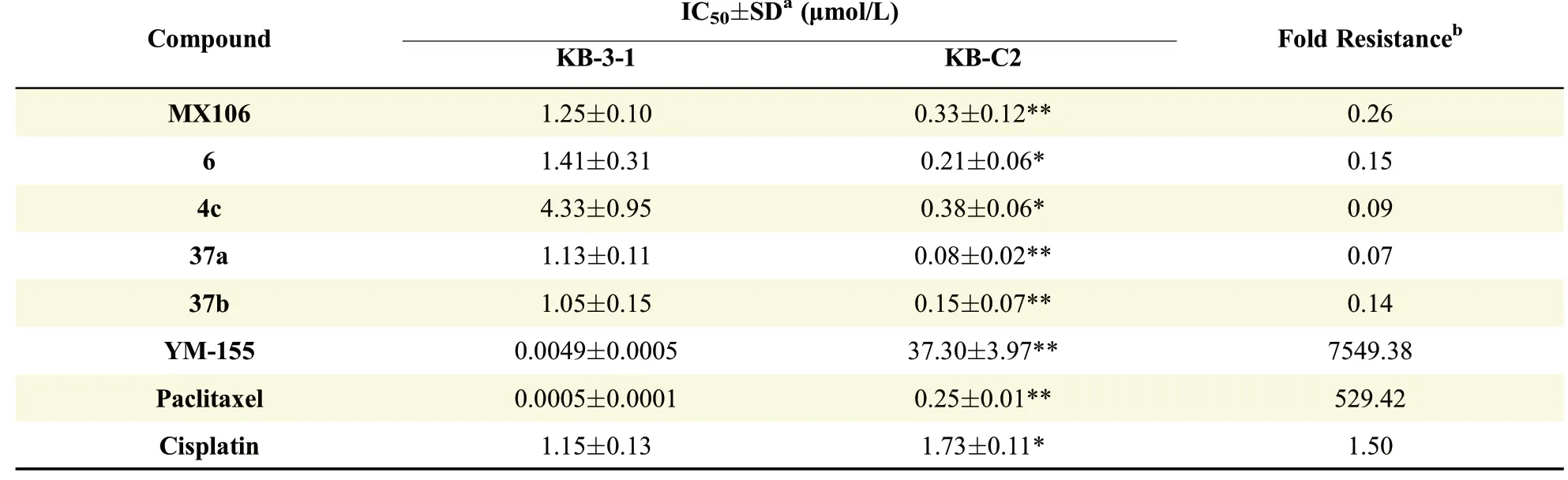
Table 3 Cytotoxic effects of f i ve analogs on parental KB-3-1 and P-gp overexpressing KB-C2 cell lines.
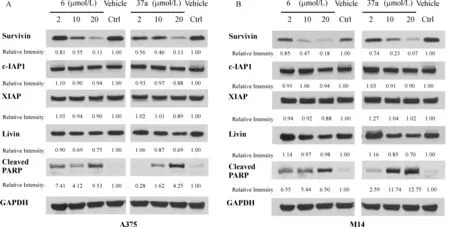
Fig.12 Western blotting of representative compounds 6 and 37a in suppressing survivin and itsclosely related IAPproteinsin A375 cells(panel A)and M14 cells(panel B).Relative band intensities represent the average of three independent replicates.In both cell lines,compound 6 and 37a selectively suppress survivin expression compared with other IAPproteins.
Mechanism of action studies
Our previous study has established that MX106 produces its antiproliferative activity by selectively inhibiting survivin expression.To determine whether the new analogs maintain their mechanisms of action,we selected two of the most potent MX106 analog,compound 6and 37a,and using Western blotanalysisto evaluate their inhibitory pro f i le against a panel of IAP proteins.
As shown in Fig.12,compound 6 and 37a,which showed comparatively potent cytotoxicity in tested cancer cell lines,indeed displayed selective and dosedependent survivin inhibition effects on human melanoma A375 and M14 cells after 24 hours treatment at concentrations from 2 to 20µmol/L.At the highest concentration tested(20µmol/L),both compound 6 and 37a effectively reduced over 80%or 90%survivin level in A375 or M14 cells,respectively.No signi f i cant inhibition effect was detected in other IAP proteins including cIAP1,XIAP,and Livin.In addition,we noticed that treatment of compound 6 or 37a signi f icantly increased the level of cleaved PARPin both cell lines,which is an important apoptosis marker and main cleavage product of the activated caspase-3.
Discussion
In this work,we performed systematic structural modi f i cations to our previously reported MX106 scaffold.SAR was determined through structural modi f i cations to the A ring,B ring,C ring,D ring and the linker of MX106 scaffold.The SAR information will provide useful directions for future development.
The most potent compound 37a had IC50value in nanomolar range in several cancer cell lines tested in thisreport.Compound 37a ismorepotent than thebest compound MX106 reported previously.
The MDR evaluation result suggests that our compounds could effectively overcome P-gp mediated MDR and surprisingly induce more potent cytotoxic effects on P-gp overexpressing cells than their parental cells.A similar f i nding reported by Ludwig,et al.showed that athiosemicarbazonederivative NSC73306 was more cytotoxic to isogenic KB cell lines with high P-gp expression than to the P-gp-negative KB-3-1 cell line[34].However,the precise mechanism of the phenomenon remains unclear.Previous studies suggested that survivin plays an important role in MDRin the presence of P-gp,and that inhibition of survivin could sensitize MDR cells to anticancer drugs[35-37].It was reported that in the KB cell line,human breast cancer MCF-7 cell line,and their corresponding P-gp overexpressing cell lines KBv200and MCF-7/ADR,neither upregulation nor downregulation of survivin could affect P-gp expression on protein level[34],but the PI3k/Akt pathway was found to be involved in P-gp associated survivin transcription activity and affected the expression of both P-gp and survivin in the same pattern in MCF-7/ADR[36].These suggested that survivin in MDR cancer cells may interact with P-gp indirectly.Although these f i ndings might be cell-type speci f i c,we could not eliminate the possibility that the compoundsin our study may achieve circumvention of P-gp-mediated MDR through a similar mechanism indirectly affecting P-gp and survivin.It isalso possible that by similar mechanism of action on P-gp,our new analogs could overcome MDRmediated by other ABC transporters.
We are currently working on to determine the activity of MX106againstother clinically relevantdrug resistance mechanisms and will report our f i ndings in the future.Mechanistically,Western blotting analysescon f i rmed that thenew compoundsmaintained thesamemodeof action as MX106 by selectively suppressing survivin expression levels.The excellent activity of those new survivin inhibitors warrants further development.
Acknowledgments
This work supported by NIH/NCI grant 1R01CA 193609-01A1.Its contents are solely the responsibility of the authors and do not necessarily represent the of f i cial views of the NIH/NCI.We thank Dr.Susan E Bates(Columbia University)for the SW620,SW620/Ad300 cell lines.We thank Dr.Shin-Ichi Akiyama(Kagoshima University,Japan)for the KB-3-1 and KBC2 cell lines.
杂志排行

THE JOURNAL OF BIOMEDICAL RESEARCH的其它文章
- Angiopoietin-like protein 3(ANGPTL3)de f i ciency and fam ilial combined hypolipidemia
- Computer-aided identi f i cation of protein targets of four polyphenols in Alzheimer’s disease(AD)and validation in a mouse ADmodel
- Melatonin and/or rowatinex attenuate streptozotocin-induced diabetic renal injury in rats
- Effects of lysophosphatidic acid on human periodontal ligament stem cells from teeth extracted from dental patients
- Effect of restoration technique on resistance to fracture of endodontically treated anterior teeth with f l ared root canals
- Maxillary denture f l ange and occlusal discrepancies of Vertex ThermoSens in comparison with conventional heat-cured denture base materials