基于CRISPR-Cas9系统的Saccharomyces cerevisiae基 因删除
2019-04-12牛潇迪李天明刘金雷杨天勇李子怡冯惠勇
牛潇迪,李天明,刘金雷,杨天勇,李子怡,冯惠勇,*
(1.河北科技大学生物科学与工程学院,河北 石家庄 050018;2.马里兰大学帕克分校文理科学学院,美国 马里兰 20742)
酿酒酵母(Saccharomyces cerevisiae)是食品级GRAS(generally regarded as safe)微生物,是酿造业、焙烤业广泛使用的微生物,也是重组蛋白高效表达的理想宿主菌之一。另外,由于其具有鲁棒性好、细胞量大、在低pH值条件(甚至pH值小于3.0)下生长良好、能耐受高浓度的底物和发酵产品具有安全性等优点,作为“细胞工厂”的底盘细胞,被广泛应用于食品添加剂、有机酸、饲料添加剂等生产[1-5]。基因组改造是提高生产性能的有效途径。常规的S. cerevisiae的基因改造方法是采用Cre-loxP系统[6],该方法利用同源重组机制采用抗生素或者营养缺陷型进行筛选,一般一次只能进行单个基因的删除;由于适用于S. cerevisiae的抗生素和营养缺陷型种类数量的限制,当进行多个基因删除时,还要进行第二次转化,借助Cre-loxP系统去除筛选标记后,才能进行;因此此方法在打靶效率、操作简便性及工作效率等方面,都有待改进提高。近年来,迅速发展完善的CRISPRCas9基因编辑技术,可以依靠向导RNA(guide RNA,gRNA)识别目标序列,利用Cas9蛋白切割序列[7-11],实施靶位点特异性切割产生DNA双链断裂,激发细胞同源重组修复机制或者非同源末端连接修复机制,无需利用筛选标记,就可实现高效的基因组的定点敲除、定点敲入和基因修饰[12-13]。CRISPR-Cas9系统相较于重组锌指核酸酶(zinc-finger nucleases,ZFNs)技术[14]和转录激活因子样效应物核酸酶(transcription activator-like effector nucleases,TALENs)基因编辑技术,有操作简单[15-17]、设计灵活、价格低廉、能实现多点编辑、可扩展性强等优点[18-20]。Church[20]和Bao Zehua[21]等先后报道了利用CRISPR-Cas9进行S. cerevisiae单倍体细胞基因组编辑的方法,但是利用该方法对S. cerevisiae双倍体细胞进行高效基因组编辑的方法还鲜见报道。
本研究通过构建Cas9和gRNA的表达质粒,在S. cerevisiae双倍体细胞中建立了CRISPR-Cas9系统,借助供体DNA(donor DNA)[22-24],实现了can1、pdc、adh3、adh2、adh1、pdc等多个单基因的高效敲除;建立了基因连续敲除和两个以上基因同时敲除的方法和程序;为在S. cerevisiae中进行大规模的基因编辑提供了方法学基础,同时也为CRISPR-Cas9技术在其他工业微生物中的应用提供了借鉴。
1 材料与方法
1.1 材料与试剂
1.1.1 菌株与质粒
本实验所用的菌株、质粒分别如表1所示。

表1 本实验所用的菌株、质粒Table 1 Strains and plasmids used in this study

续表1
1.1.2 引物与试剂
本实验所用引物如表2所示。

表2 本实验所用引物Table 2 PCR primers used in this study
构建及验证所用聚合酶链式反应(polymerase chain reaction,PCR)引物由生工生物工程(上海)股份有限公司合成;FastPfuDNA聚合酶、High Fidelity DNA聚合酶 北京全式金生物技术有限公司;限制性内切酶KpnI、XhoI、SacI、NotI NEB(北京)公司;T4 DNA ligse 大连宝生物工程公司;琼脂糖凝胶DNA回收试剂盒、质粒小提中量试剂盒、超薄DNA产物纯化试剂盒和基因组提取试剂盒 天根生化科技(北京)有限公司。
转化过程中用到10×TE、1×TE、10×LiAc、1×LiAc(0.1 mol/L),1×LiAc/0.5×TE为10 mL 10×LiAc和5 mL 10×TE用去离子水定容100 mL,1×LiAc/40%聚乙二醇(polyethylene glycol,PEG)3350/1×TE(现用现混)为100 μL 10×LiAc+100 μL 10×TE+800 μL 50% PEG3350混合。
1.1.3 培养基
LB(Luria-Bertani)培养基:胰蛋白胨10 g/L、酵母提取物5 g/L、NaCl 10 g/L,调pH值为7.4左右,需要时加入卡那霉素至终质量浓度0.05g/L。固体培养基则添加琼脂粉至20 g/L。
SOC(super optimal broth with catabolite repression)培养基:2 g/100 mL胰蛋白胨、0.5 g/100 mL酵母粉、0.05 g/100 mL氯化钠、2.5 mmol/L氯化钾、10 mmol/L氯化镁、20 mmol/L葡萄糖。
酵母浸出粉胨葡萄糖(yeast extract peptone dextrose,YPD)培养基:1 g/100 mL酵母膏、2 g/100 mL 蛋白胨、2 g/100 mL葡萄糖,若制固体培养基,加入2 g/100 mL琼脂粉。
选择(synthetic dropout,SD)培养基:无氨基酵母氮源(yeast nitrogen base,YNB)培养基(成分见Solarbio YNB培养基产品介绍)6.7 g,另取葡萄糖5 g加热溶解于100 mL蒸馏水中,制成10 倍溶液,过滤除菌。使用时,取0.5 mL 10 倍溶液加入4.5 mL无菌蒸馏水中,备用。根据不同的筛选标记添加不同的氨基酸。
1.2 仪器与设备
Finnpipette移液枪 美国Thermo公司;SW-CJ型超净工作台 苏净集团安泰公司;PCR仪 美国Bio-Rad公司;Jl003A型电子天平 常熟双杰测试仪器厂;HWS型培养箱 宁波江南仪器厂;F1-F2凝胶成像仪上海天能科技有限公司;Neofuge23R冷冻离心机 力新仪器(上海)有限公司;pH211酸度计 意大利Hanna公司;HYG型摇床 上海欣蕊自动化设备有限公司;PoweWave HT酶标仪 美国伯腾仪器有限公司;DZKW-D水浴锅 河北黄骅航天仪器厂;BCD-191W/HC冰箱 上海海尔股份有限公司;G80W23YCsL-03(RO)微波炉 上海申安医疗器械厂。
1.3 方法
1.3.1 菌株培养
本研究所用的S. cerevisiae菌株为4 种氨基酸缺陷的双倍体菌株,缺陷型为Trp-、Leu-、Ura-、His-。亲本S. cerevisiae菌株Δura3Δtrp1Δleu2Δhis3(下文均称为WT)转化前在YPD培养基,30 ℃、180 r/min条件下培养。转化后的筛选培养基采用缺乏相应质粒的筛选标记的筛选培养基,30 ℃、200 r/min条件下培养。
1.3.2 CRISPR-Cas9系统的构建

图1 pRS424-TRP-cas9构建示意图Fig. 1 Schematic illustration of construction of pRS424-TRP-cas9
CRISPR-Cas9系统需要Cas9核酸酶和gRNA的共同作用才能发挥基因编辑作用,需要构建一个能够表达cas9的质粒,转化进酵母菌中使其表达Cas9核酸酶[25-30]。表达Cas9核酸酶的质粒构建方法如下:首先以实验室保存质粒pMD18-cas9为模板,通过引物cas9-F、cas9-R利用PCR技术扩增出cas9基因片段;以S. cerevisiae288c野生型菌株基因组为模板,通过引物cas9-tef-F、cas9-tef-R利用PCR技术扩增出tef启动子;以S. cerevisiae288c野生型菌株基因组为模板,通过引物cas9-cyc-F、cas9-cyc-R利用PCR技术扩增出cyc1终止子。为使在细胞质中表达的Cas9蛋白质能够透过核膜到达细胞核内参与DNA剪切,在Cas9表达质粒中添加一段表达核定位信号(nuclear localization signal,NLS)的基因序列,通过在tef启动子基础上利用延伸PCR扩增得到,将所得片段利用Overlapping PCR技术融合得到目的片段,凝胶纯化目的片段后与pRS424-TRP空质粒利用KpnI和XhoI进行双酶切,目的片段与pRS424-TRP空质粒的酶切产物通过超薄DNA产物回收试剂盒回收,利用T4连接酶过夜连接。产物转化E.coliTransT1感受态中,转化子提取质粒,经酶切验证后获得Cas9表达质粒pRS424-TRP-cas9。构建示意图如图1所示。质粒pRS424-TRP带有色氨酸(Trp)表达基因,WT为Δura3Δtrp1Δleu2Δhis3基因缺陷型,将pRS424-TRP质粒转化进WT细胞后,可利用Trp营养缺陷的培养基筛选。
被敲除基因的靶位点gRNA表达质粒构建及其donor DNA的设计:以敲除can1基因的打靶质粒为例,参照NCBI中S. cerevisiae模式菌株S. cerevisiae288c的基因组信息,选取编码精氨酸透酶的基因can1为靶点基因,利用软件找到cas9可以识别的间隔序列前体旁基序(protospacer adjacent motif,PAM)位点(5-NGG或NAG结构),通过引物can1-gRNA-1F、can1-gRNA-1R利用PCR扩增出SNR52启动子、HINDLE,通过引物can1-gRNA-2F、can1-gRNA-2R利用PCR扩增出SUP43FLANK,利用Overlapping PCR技术融合启动子SNR52、20 nt can1靶点序列、HINDLE及SUP43FLANK,将所得片段纯化后与pESCG-LEU质粒骨架利用SacI和NotI进行双酶切,目的片段与pESCGLEU空质粒的酶切产物通过超薄DNA产物回收试剂盒回收,利用T4连接酶过夜连接,产物转化E. coli TransT1感受态中,转化子提取质粒,经酶切验证后获得打靶质粒pESCG-LEU-gRNA-can1。构建示意图如图2所示。

图2 pESCG-LEU-gRNA-can1构建示意图Fig. 2 Schematic illustration of construction of pESCG-LEU-gRNA-can1
设计引物融合靶位点上下游的一段同源序列,以S.cerevisiae 288c基因组为模板,通过PCR技术扩增出donor DNA。构建示意图如图3所示。
本实验还成功敲除了adh1、adh2、adh3、pdc、pdh等多个基因,其打靶质粒详见表1。本实验所用的菌株、质粒。打靶质粒的构建方法和donor DNA的构建方法与前文所述类似,此外,一次转化两个打靶质粒pRS423-HIS-gRNA-qadh5及pESCG-LEU-gRNA-lip的构建方法和donor DNA的构建方法也与之类似。
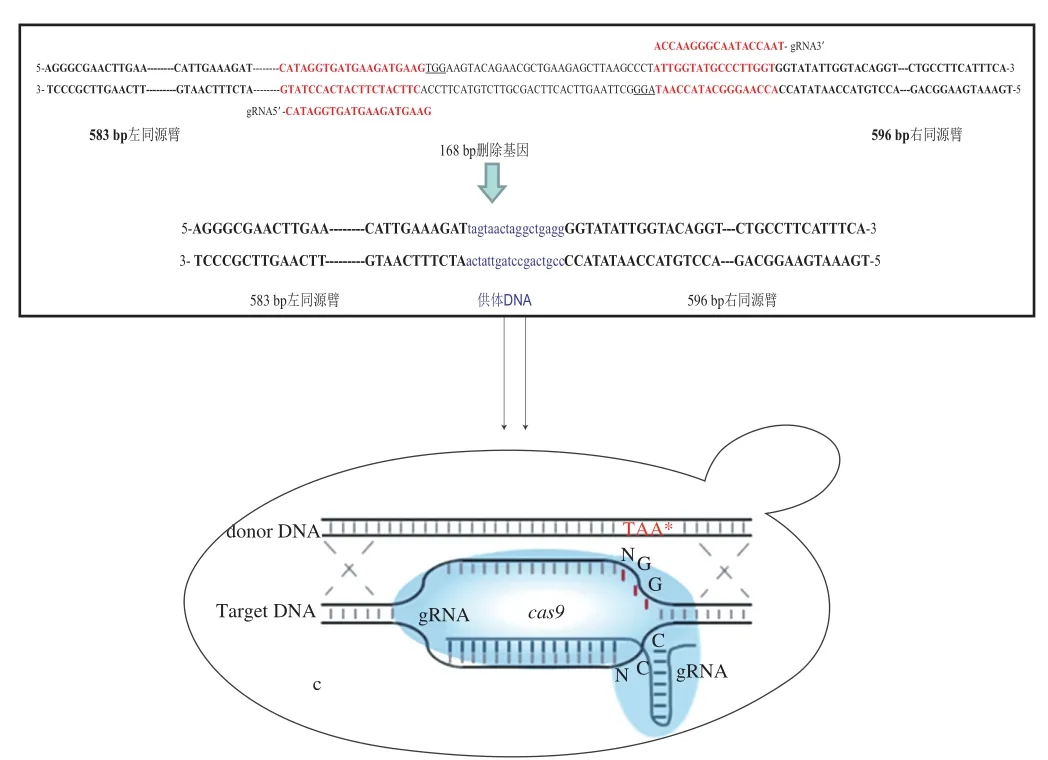
图3 donor DNA构建示意图Fig. 3 Schematic illustration of construction of donor DNA
1.3.3 S. cerevisiae感受态的制备及转化
S. cerevisiae感受态的制备方法参考文献[31]酵母高效转化,稍作修改。将过夜培养的菌株WT以10%体积比接种于新鲜的YPD培养基,30 ℃、180 r/min培养3~4 h,当OD600nm达到0.5~1时,利用冷冻离心机在4 ℃条件下收集菌体,弃去上清液,加1 mL 1×TE重悬,弃去上清液,30 μL 1×LiAc/0.5×TE重悬,之后室温静置10 min,加入1 μg pRS424-TRP-cas9,1 μg打靶质粒,10 µL donor DNA和10 µL ss-DNA,加入250 μL 1×LiAc/40% PEG3350/1×TE,混合均匀,30 ℃孵育30 min,加入30 μL二甲基亚砜,混合均匀,42 ℃热击7 min,4 000 r/min离心2 min,弃去上清液,加入1 mL 1×TE重悬漂洗,最后离心收集菌体涂布筛选平板。30 ℃培养约72 h后,挑选固体培养基平板上长出的第3批较小的单菌落进行筛选验证。
1.3.4 can1靶点的筛选验证
由于can1基因编码一种质膜精氨酸(Arg)通透酶,所以在敲除can1基因的实验中,将转化进打靶质粒的酵母感受态涂布在添加精氨酸的培养基上。待长出转化子后挑取单菌落影印在含有刀豆素(一种有毒精氨酸类似物)和不含刀豆素的培养基上,若can1基因被敲除,则菌在刀豆板上生长,若没有敲除can1基因,则菌在刀豆板上不长。由此可以挑选出正确的转化子。
之后设计验证引物进行分子验证,敲除基因不能扩增出来,敲入基因能扩增出来的为阳性转化子。
1.3.5 其他基因敲除的筛选验证
挑选培养72 h后第3批长出的较小的菌落扩菌到相同筛选标记的固体培养基平板上,第2天振荡裂解进行菌落PCR验证,确认打靶细胞经过同源重组正确地整合到宿主细胞中。将验证正确的菌株再次扩菌,提取基因组进一步验证确认。
1.4 数据及图像处理
基因敲除效率的计算方法:将PCR验证正确的阳性转化子的个数与所有被验证转化子的比例作为这一基因的敲除效率。凝胶成像图利用visio软件处理并标注。
2 结果与分析
2.1 Cas9系统的构建
2.1.1 pRS424-TRP-cas9质粒的构建
以pMD18-cas9的基因组为模板经PCR扩增出cas9基因片段;以S. cerevisiae 288c野生型菌株基因组为模板,利用PCR技术扩增出tef启动子,cyc1终止子,NLS在tef启动子基础上通过延伸PCR扩增得到,利用Overlapping PCR技术融合得到目的片段,将目的片段与空质粒pRS424-TRP双酶切后连接,转化到E. coli,转化子提取质粒进行酶切验证,获得5 616 bp载体片段和4 860 bp目的片段2 条条带,结果如图4所示,质粒pRS424-TRP-cas9构建成功。
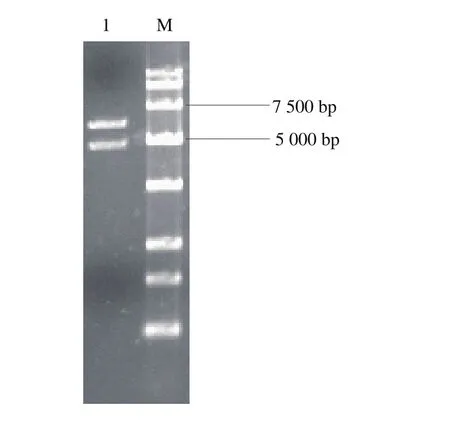
图4 pRS424-TRP-cas9酶切验证Fig. 4 Identification of pRS424-TRP-cas9 by double enzymatic digestion
2.1.2can1基因gRNA打靶质粒的构建
以S. cerevisiae288c基因组为模板,利用PCR扩增技术得到can1靶点基因、SNR52启动子、HINDLE及SUP43FLANK,利用Overlapping PCR技术融合得到目的片段,将目的片段与空质粒pESCG-LEU双酶切后连接,转化到大肠杆菌,转化子提取质粒进行酶切验证,得到400 bp大小目的片段和7 200 bp大小载体片段,如图5所示,质粒pESCG-LEU-gRNA-can1构建成功。
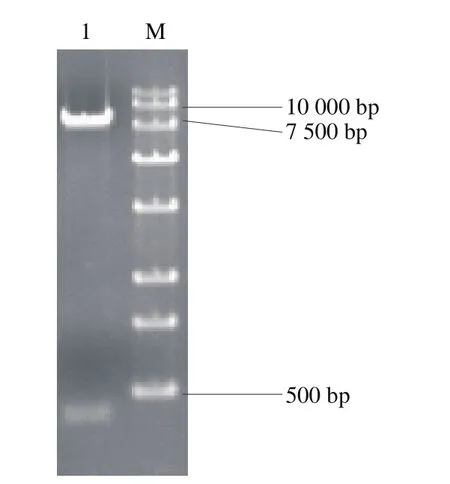
图5 打靶质粒pESCG-LEU-gRNA-can1酶切验证Fig. 5 Identification of target plasmid pESCG-LEU-gRNA-can1 by double enzymatic digestion
donor DNA的构建:以S. cerevisiae288c基因组为模板,通过引物融合靶位点上下游的一段同源序列,得到一段大小为1 450 bp的基因片段,凝胶回收备用。
2.2 can1靶点基因敲除的结果
将表达Cas9核酸酶的表达质粒pRS424-TRP-cas9、打靶质粒pESCG-LEU-gRNA-can1以及donor DNA共同转化进S. cerevisiae感受态细胞内,Cas9核酸酶与gRNA共同作用实现细胞体内重组。将单菌落用0.9%生理盐水稀释,在含有刀豆素(CAN+)和不含刀豆素(CAN-)的培养基上影印,含刀豆的板上长的即为阳性转化子。如图6所示,统计得到阳性抗刀豆氨酸的突变子大约22%。将影印结果中阳性转化子的对应菌液振荡裂解做PCR菌落验证,根据待敲除基因can1以及donor DNA设计敲除验证引物,验证被敲除基因的引物为YZQCF/YZQCR,其中,F为被敲除基因的23 bp大小的基因序列(AGGATGGCATAGGTGATGAAGAT,GenBank号为NC_001133.9),R为下同源臂下游染色体27 bp大小基因序列(GCAGTGATACCAACTAGTTCAGTACCT,G e n B a n k号为N C_0 0 1 1 3 3.9)。验证敲入基因的引物为Y Z Q R F/Y Z Q R R,其中F为d o n o r D N A中含有终止密码子的2 1 b p序列(AATAACGGCAAACAGCAAAGG,GenBank号为NC_001133.9),R为下同源臂下游染色体27 bp大小基因序列(GCAGTGATACCAACTAGTTCAGTACCT,GenBank号为NC_001133.9)分别以can1-/WT菌株与WT菌株的基因组为模板,进行PCR验证,结果如图7所示,敲除基因没有扩增出来,敲入基因扩增出来,大小为750 bp为阳性转化子,22 个中有4 个为基因编辑的转化子。生理突变株和分子验证的突变株数量不一致的原因,可能是S. cerevisiae为真核细胞,存在着非同源性末端接合,虽然没有借助同源重组也得到了can1基因改变的突变株。

图6 can1基因敲除影印结果Fig. 6 Photographic results of knocking-down can1 gene

图7 can1缺失基因PCR验证Fig. 7 Identification of can1 deletion by PCR
2.3 其他单基因敲除的结果
为验证建立的CRISPR-Cas9系统在S. cerevisiae中进行基因编辑的有效性,本研究分别构建pdc、adh3、adh2、adh1、pdh基因gRNA的表达质粒及相应的打靶donor DNA(其他基因敲除表达质粒的构建结果略)分别转化S. cerevisiae感受态细胞,Cas9核酸酶与gRNA共同作用实现细胞基因编辑。待培养72 h后,挑取最小一批的单菌落扩菌,PCR验证,汇总实验结果,总体基因编辑效率如表3所示。基因pdc敲除效率为4/48;adh3敲除效率为3/48;adh2敲除效率为1/48;adh1敲除效率为3/28;pdh敲除效率为1/16。

表3 各基因敲除效率Table 3 Gene knockdown efficiency
2.4 基因连续敲除的结果
为在敲除掉某一个基因后能够快速顺利地敲除下一个基因,采用筛选标记交替使用的方法:敲除第1个基因的gRNA构建在Leu作为筛选标记的质粒上,第2个敲除的基因构建在His作为筛选标记的质粒上,第1个基因敲除后,使用添加第1种筛选标记的Leu的培养基,继续进行第2个基因的敲除操作,这样可以在进行敲除基因的同时,丢掉第1个基因的pESCG-LEU-gRNA质粒,以便进行第3个基因敲除操作,这样交替进行,提高了多个基因敲除的操作效率,简化了操作步骤。连续基因敲除流程如图8所示。采用上述方法,本研究在pdc基因敲除后,继续敲除了adh3基因,又继续敲除了adh2,敲除3 个基因整个操作过程为17 d。根据待敲除基因以及donor DNA设计敲除验证引物,分别以待验证的敲除基因菌株与WT菌株的基因组为模板,进行PCR验证,第1个基因pdc敲除的验证结果如图9所示,敲入基因扩增出来,大小为560 bp为阳性转化子;第2个基因adh3敲除的验证结果如图10所示,敲入基因扩增出来,大小为850 bp为阳性转化子;第3个基因adh2敲除的验证结果如图11所示,敲除基因扩增不出来,敲入基因扩增出来,大小为570 bp为阳性转化子。
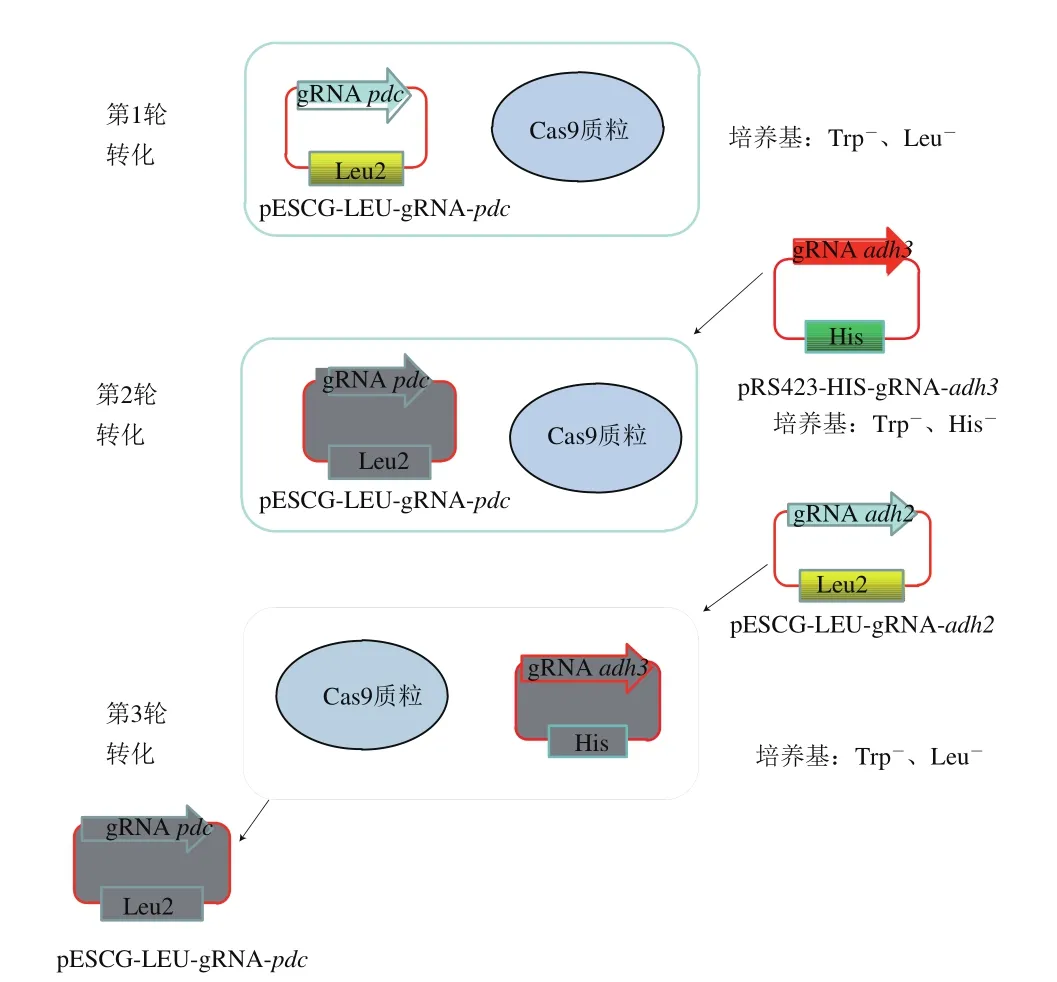
图8 连续基因敲除流程图Fig. 8 Flow chart of sequential knockout
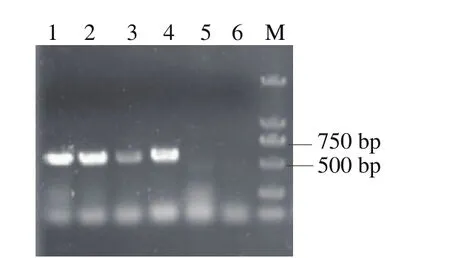
图9 pdc缺失基因PCR验证Fig. 9 Identification of pdc deletion by PCR

图10 adh3缺失基因PCR验证Fig. 10 Identi fi cation of adh3 deletion by PCR

图11 adh2缺失基因PCR验证Fig. 11 Identification of adh2 deletion by PCR
2.5 双基因敲除的筛选验证结果
为进一步提高CRISPR-Cas9系统的基因删除效率,本研究将pRS424-TRP-cas9、gRNA表达质粒pRS423-HIS-gRNA-adh5、pESCG-LEU-gRNA-lip以及donor DNA共同转化进S. cerevisiae感受态细胞内,Cas9核酸酶与gRNA共同作用实现细胞体内重组。待培养72 h后挑取最小的一批单菌落扩菌验证,根据待敲除基因lip以及donor DNA设计敲除验证引物,分别以lip-/WT菌株与WT菌株的基因组为模板,进行PCR验证,结果如图12所示,敲除基因没有扩增出来,敲入基因扩增出来,大小为750 bp为阳性转化子,统计得到转化效率为9/32。根据待敲除基因adh5以及donor DNA设计敲除验证引物,分别以adh5-/WT菌株与WT菌株的基因组为模板,进行PCR验证,结果如图13所示,敲除基因没有扩增出来,敲入基因扩增出来,大小为1 400 bp为阳性转化子,统计得到转化效率为10/32。

图12 lip缺失基因PCR验证Fig. 12 Identification of lip deletion by PCR
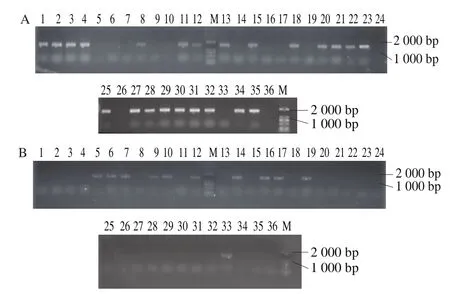
图13 adh5缺失基因PCR验证Fig. 13 Identification of adh5 deletion by PCR
3 讨 论
酵母是一种单细胞真核生物,其清晰的遗传背景、简易的分子遗传操作、可控的发酵过程及低廉的发酵生产成本,在外源蛋白表达生产及作为细胞工厂生产生物能源和化学品中发挥了极大的优势。传统的酵母基因编辑通常采用同源片段打靶抗性筛选的方法,往往因抗性标记在染色体上的整合而无法重复使用和无法去除,影响其在工业上的应用。借助酵母的营养缺陷底盘细胞和Cre重组酶的酵母基因修饰改造系统[6],虽然可以实现基因组的无痕改造,但需要两次转化,而且反向筛选假阳性多,造成操作繁琐、周期长,也影响了酵母基因编辑的大规模实施。本研究采用CRISPR-Cas9基因编辑技术,只需构建gRNA和Cas9表达质粒,共转化S. cerevisiae细胞,造成靶位点特异性切割,大大提高了供体DNA的重组效率,无需抗性筛选标记的筛选,就可高效率进行基因组基因的删除、插入及修饰。与锌指核酸酶ZFN[15-16]和转录激活因子效应物核酶TALEN[17]的基因编辑技术相比,CRISPR-Cas9系统可以有效地实现多个基因的编辑,在应用中更低成本、更简便、更高效[18-19],是解决酵母基因编辑上述瓶颈问题的有效工具[32-34]。
酵母工业菌株都是双倍体,对于双倍体S. cerevisiae基因编辑的难度大于单倍体细胞。Church等[20]第一次利用CRISPR-Cas9基因编辑技术将单倍体S. cerevisiae中的can1基因敲除,效率为0.7%。Bao Zehua等[21]利用CRISPR-Cas9系统一次转化单倍体S. cerevisiae后,通过液体长程培养6 d后再进行涂板筛选,可以同时敲除3 个基因。两位研究者,基因编辑的均是单倍体S. cerevisiae细胞,采用上述方法必须将双倍体酵母细胞分离为单倍体,基因修饰后再复性为双倍体,操作繁琐。本研究借鉴其研究,将CRISPR-Cas9基因编辑技术拓展应用到双倍体S. cerevisiae细胞的基因定向改造中,成功地将can1、pdc、adh3、adh2、adh1、pdh基因敲除,这种针对工业双倍体菌株的基因编辑方法还鲜见报道。本研究还探索高效率连续敲除基因的方法流程,完全敲除3 个基因整个操作过程为17 d。利用2 个筛选标记的gRNA一次转化同时删除两个lip和adh5基因也成功实现。本研究也探索了3 个基因同时敲除的方法,但由于2 个gRNA表达质粒及Cas9表达质粒采用4 种筛选标记筛选,而且3 种供体DNA同时转化,影响转化DNA的通量,转化子较少,造成阳性突变株很少或没有(数据略)。在以后研究中可改进为:采用不同基因的靶点gRNA构建在同一个表达质粒上,就可解决转化子少的问题,实现多个基因的同时删除及修饰。
