肌萎缩侧索硬化症患者骨骼肌神经末梢中磷酸化TAR DNA结合蛋白43和泛素的表达
2019-04-03周乐波王雪晶丁雪冰姜晓懿滕军放
周乐波,王雪晶,丁雪冰,姜晓懿,滕军放
1)郑州大学第一附属医院神经内科 郑州 450052 2)河南省高等学校临床医学重点学科开放实验室 郑州 450052
肌萎缩侧索硬化症(amyotrophic lateral sclerosis, ALS)是一种进展性、致死性神经系统变性疾病,以锥体束、脑干和脊髓前角中选择性的运动神经元变性为特征。临床上较为少见,主要表现为进行性加重的骨骼肌萎缩、无力、肌束颤动、延髓麻痹及锥体束征[1]。ALS发病率约为1.5/10万,病情进展较快,目前尚无有效的治疗手段,患者生存质量较差,预后不良,大部分生存期只有3~5 a[2]。运动神经元及神经胶质细胞中TAR DNA结合蛋白43(TAR DNA-binding protein 43,TDP-43)聚集物(包涵体)是ALS的特异性病理特征,TDP-43包涵体呈泛素(ubiquitin,Ub)阳性,无法正常降解[3]。既往研究[4-7]描述了ALS患者萎缩肌肉的电生理学特点,目前对于ALS患者肌肉组织中异常蛋白聚集物的研究仍较少。为此,该研究通过肌肉活检术,检测ALS患者骨骼肌组织及其神经末梢中磷酸化TDP-43(pTDP-43)和Ub的表达情况,探讨早期肌肉组织受累在ALS发病中的作用。
1 对象与方法
1.1研究对象收集2012年3月至2018年6月在郑州大学第一附属医院神经内科就诊的ALS患者30例, 均符合1999年修订的Airlie House诊断标准[8]及2008年补充的Awaji-Shima标准[9],无其他神经系统疾病。22例对照为在该院外科接受手术治疗的非神经系统退行性疾病及其他慢性疾病患者。本研究通过郑州大学及郑州大学第一附属医院伦理委员会批准,获得所有受试者和(或)其家属知情同意并签署骨骼肌活检知情同意书。对所有受试者进行全面的病史采集,包括年龄、性别、症状、起病部位、病情进展程度、肌电图、家族史、生活环境、工作性质、吸烟饮酒史、中毒史、外伤史、神经系统病变危险因素接触史、其他慢性疾病史等。30例ALS患者均经随访、体格检查及神经电生理检查确诊,男19例,女11例;年龄24~80(54.7±13.4)岁,发病至确诊时间5~20个月;以单上肢起病者1例,双上肢3例,单下肢4例,双下肢5例,四肢8例,偏侧肢体2例,以延髓麻痹症状起病者3例,延髓麻痹+上肢3例,延髓麻痹+上肢+下肢1例;肌电图均提示神经源性损害,5例有家族史。22例对照均排除神经系统退行性疾病,男13例,女9例,年龄28~72(48.5±13.0)岁。
1.2肌肉组织取材选取受试者一侧上臂,分别于肱二头肌、肱三头肌、三角肌对应处用记号笔做好切口标记,碘伏溶液常规消毒,铺孔巾,20 g/L利多卡因于定位处做逐层浸润麻醉后,左手固定皮肤,右手持手术刀在拟取材处垂直进刀,沿肌纤维纵行方向分别做约3 cm长的切口,钝性分离,充分暴露肌肉组织,用血管钳游离肌束,切取肌束(长0.5~1.0 cm,直径0.3~0.5 cm),平均分为两部分,分别置于40 g/L多聚甲醛中固定及液氮低温冻存。取材完成后压迫止血、清理创面、缝合切口、消毒、无菌纱布加压包扎,8 d后拆线。
1.3肌肉组织HE及酶组织化学染色取出浸泡于40 g/L多聚甲醛内的肌肉组织标本,常规梯度乙醇脱水,经石蜡包埋制成蜡块,用石蜡切片机连续切片,厚度4 μm。取出低温冻存的肌肉组织,用冰冻切片机连续切片,厚度15 μm。
1.3.1 HE染色 苏木精液10 min,流水洗,10 g/L盐酸乙醇分化10 s;10 g/L伊红0.5~1.0 min,流水洗,梯度乙醇脱水,二甲苯透明,封片。显微镜下观察肌纤维大小、形态、结构。
1.3.2 ATP酶染色 常规ATP酶染色均在室温下进行,准备0.01 mol/L巴比妥钠4 mL、0.18 mol/L氯化钙4 mL、蒸馏水12 mL;标定酸、碱之后倒入染色小缸,将切片分别放入3种不同pH值(分别为10.40、4.65和4.30)的预孵育液内,室温下10 min;0.18 mol/L氯化钙10 min内洗3次,20 g/L氯化钴3 min,0.01 mol/L 巴比妥钠洗3次,10 g/L硫化铵30 s,水洗,梯度乙醇脱水,二甲苯透明,封片。显微镜下观察肌纤维类型、分布等。
1.3.3 还原型辅酶Ⅰ-四氮唑还原酶(NADH-TR)染色 配制反应液:将8 mg还原型烟酰胺腺嘌呤二核苷酸、10 mg硝基四唑氮蓝、10 mL 0.2 mol Tris-HCl缓冲液(pH 7.4)混匀后加入1 mL二甲基亚砜,然后用玻璃棒搅拌2~3 min,过滤后使用。将切片于反应液内37 ℃孵育35 min,丙酮清洗,蒸馏水冲洗,自然干燥,封片。显微镜下观察肌纤维大小、形态、结构及类型、分布等,数码相机拍照。
1.4神经末梢中pTDP-43和Ub表达的免疫组化检测石蜡切片烤片,二甲苯脱蜡,梯度乙醇脱水,H2O2去离子水消除内源性过氧化物酶活性,置于枸橼酸钠缓冲液中行高温高压抗原修复5 min。胰蛋白酶消化,PBS洗涤,滴加正常山羊血清封闭液,随后滴加稀释的一抗工作液(鼠抗pTDP-43抗体购自 Millipore 公司,按1∶300稀释;鼠抗Ub抗体购自Santa Cruz公司,按1∶500稀释),4 ℃ 过夜。室温下复温,PBS洗涤3遍。滴加辣根过氧化物酶标记的二抗工作液(羊抗鼠IgG,购自博奥森公司),孵育30 min,PBS洗涤3遍。DAB显色3 min,梯度乙醇脱水,二甲苯透明,封片。显微镜下观察肌细胞形态及着色情况,数码相机拍照。若胞质和胞核有棕褐色或棕黄色、粗糙颗粒状着色则记为阳性染色,计算阳性表达率。
1.5统计学处理采用SPSS 21.0进行数据分析,两组肌肉组织中 pTDP-43和Ub阳性表达率的比较均采用Fisher精确概率法,检验水准α=0.05。
2 结果
2.1两组骨骼肌肌肉组织HE及酶组织化学染色结果镜下可见ALS患者肌肉组织肌纤维大小不等,散在萎缩的小角形纤维,小群萎缩、大组状萎缩纤维,肌膜核增多,ATP酶染色可见两型肌纤维萎缩、同型群组化现象,NADH-TR染色可见靶纤维等特征性病理表现;对照组无上述表现(图1)。
2.2两组骨骼肌神经末梢中pTDP-43和Ub阳性表达率的比较镜下可见ALS患者骨骼肌神经末梢中散在分布pTDP-43及Ub阳性染色细胞(图2、3)。22例对照肌肉组织中未检出pTDP-43及Ub表达;14例(46.7%)ALS患者pTDP-43阳性表达,18例(60.0%)Ub阳性表达;两组比较,P均<0.001。
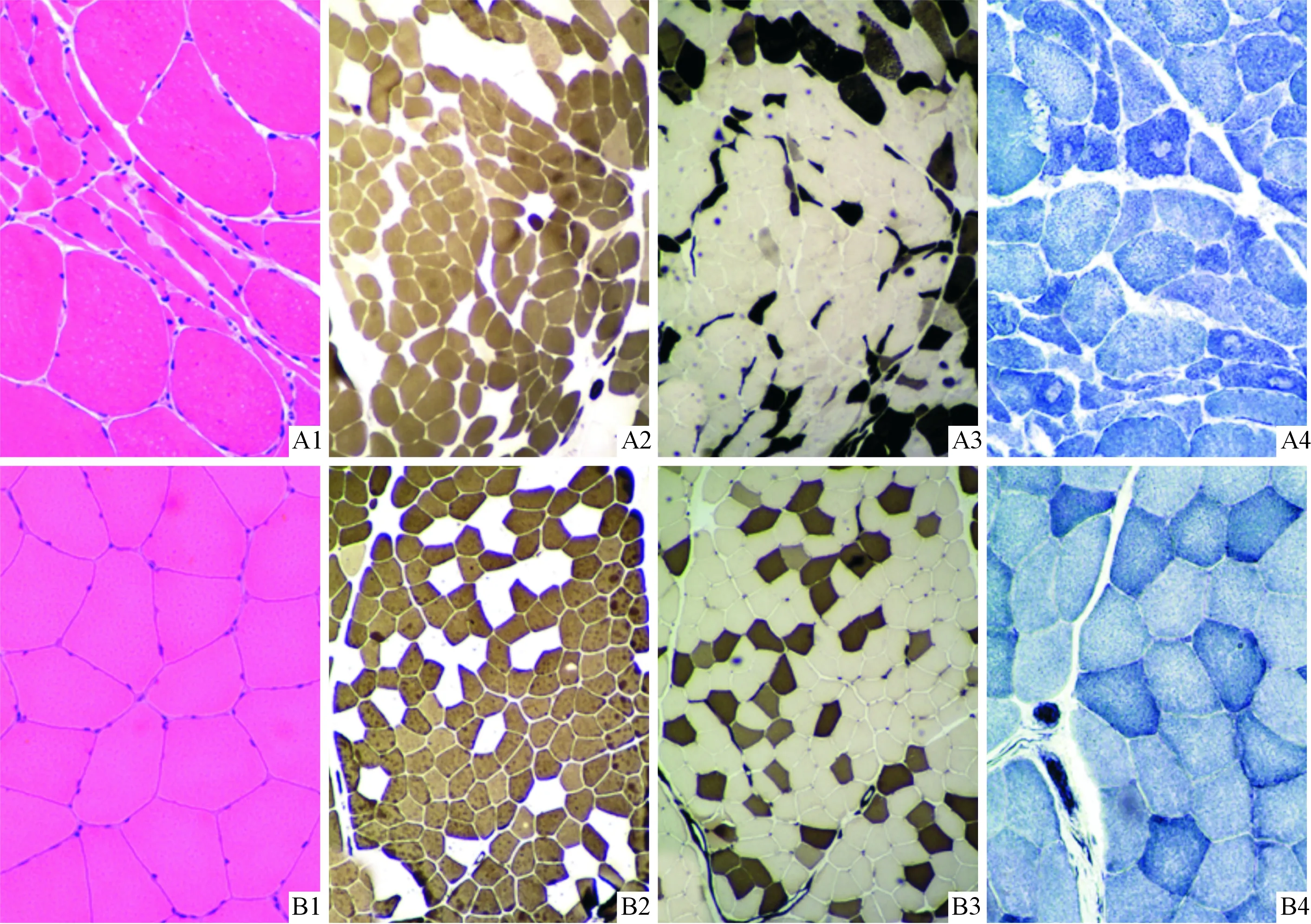
A:ALS组;B:对照组;1:HE染色(×100);2、3:ATP酶染色(×40);4:NADH-TR染色(×100)
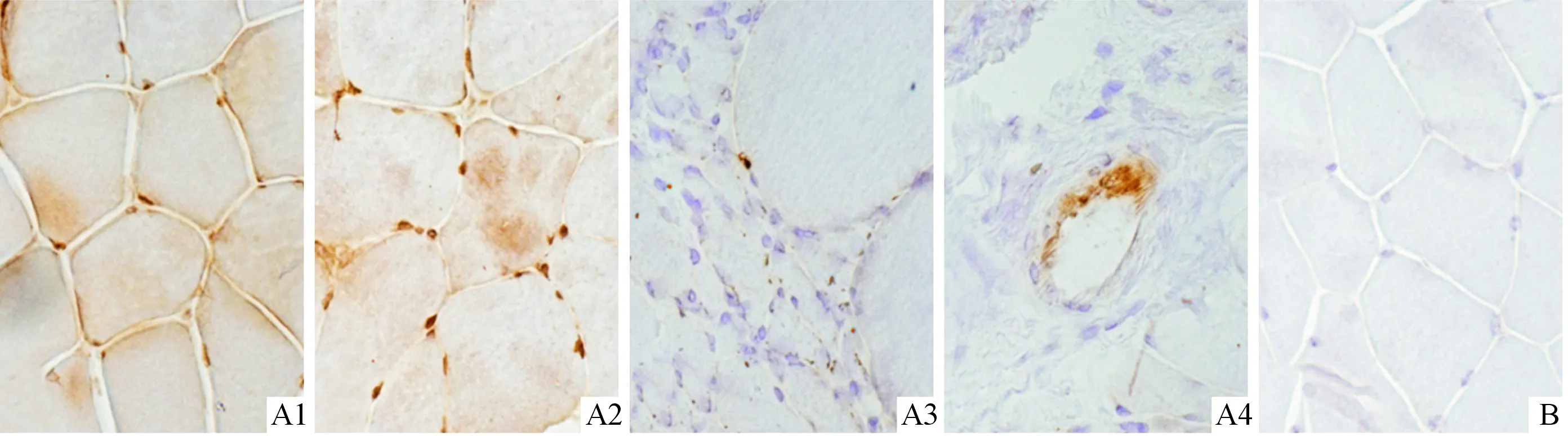
A:ALS组;B:对照组;1、2:肌纤维;3、4:骨骼肌神经末梢
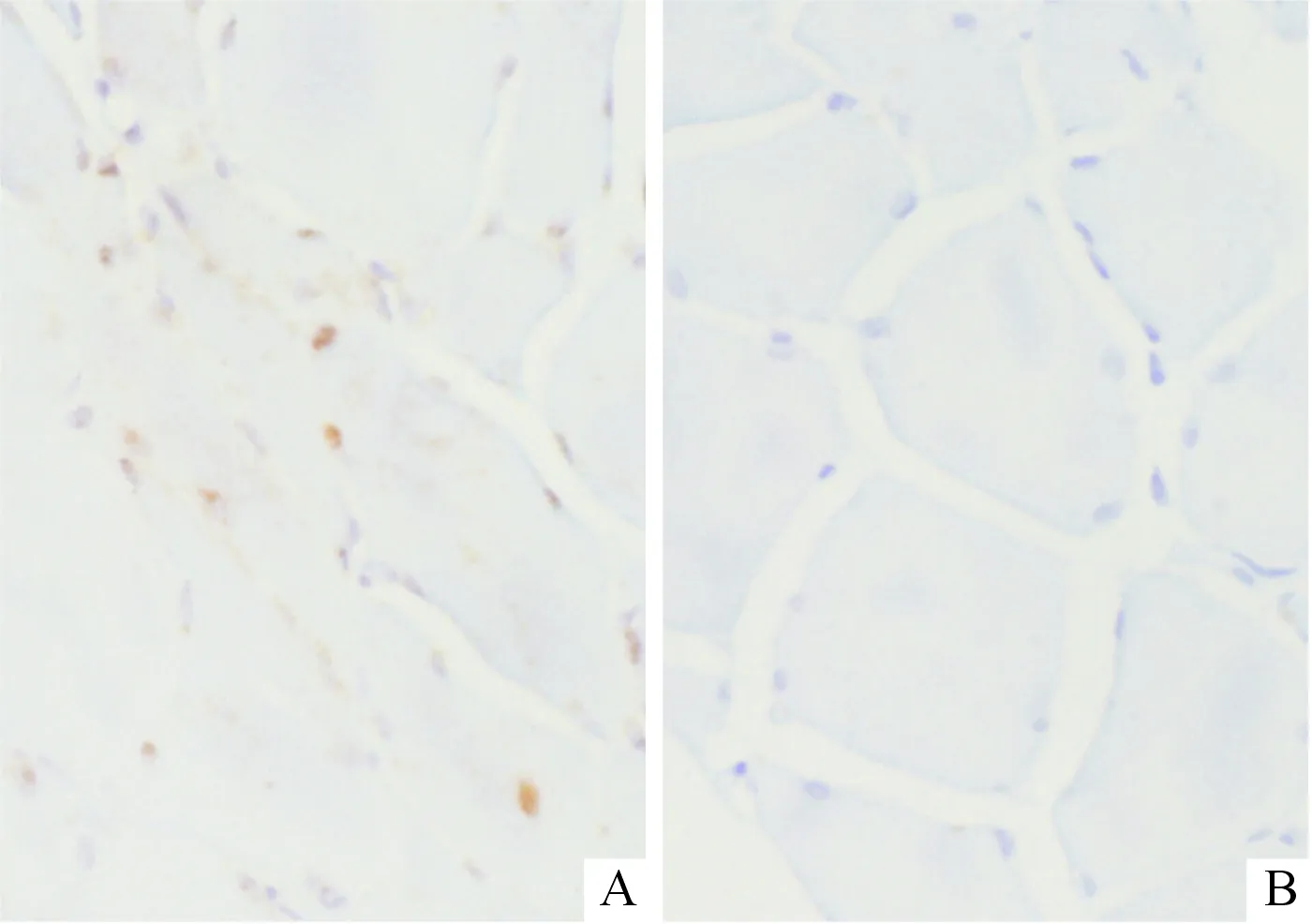
A:ALS组;B:对照组
3 讨论
ALS是一种累及锥体束、脑干运动神经核及脊髓前角细胞的慢性进行性变性疾病,其病因及发病机制不明。ALS的病理学标志是运动神经元及神经胶质细胞胞质内散在分布的Ub阳性异常束状蛋白聚集、颗粒样物质和球状包涵体,TDP-43是其核心成分[3]。以ALS为代表的TDP-43 蛋白沉积为病理特征的一组疾病统称为TDP-43蛋白病[10]。Ub阳性包涵体是神经变性疾病的病理特征之一,如阿尔茨海默病的神经纤维缠结、帕金森病的路易小体及亨廷顿病的亨廷顿蛋白包涵体均为Ub免疫反应阳性。在ALS患者的脊髓、脑干、运动皮层的运动神经元及海马神经元中均分布有Ub阳性包涵体。既往有研究[11]表明,Ub-蛋白酶体系统(Ub-proteasome system, UPS)可能与ALS发病机制有关,UPS异常可导致ALS患者TDP-43蛋白异常聚集。
目前认为,ALS的发病源于运动神经元的损害,继发产生失神经性肌肉萎缩。电生理学研究[7]证实,ALS患者的肌肉表现为异常的自发活动、运动单位募集减少、运动单位数量减少、传导速度减慢、复合肌肉动作电位两侧不对称、慢性重复刺激衰减等。病理组织学研究[6]证实,ALS患者的肌肉组织具有多重病理学改变,包括靶纤维、群组化现象,纤维坏死和炎症,DNA片段化与促凋亡蛋白的表达等。然而,关于ALS患者肌肉组织中pTDP-43沉积的病理研究较少。有学者[12]对30例ALS患者进行股四头肌活检术,未检测到pTDP-43沉积。另有学者[13]于ALS患者腓肠肌中检测出Ub结合蛋白P62阳性而pTDP-43阴性的蛋白包涵体。研究[6]发现,33.3%的ALS患者肌肉组织中可检测出pTDP-43和P62阳性包涵体,尤以轴向骨骼肌(椎旁肌、膈肌)为甚。本研究中作者检测了ALS患者骨骼肌神经末梢中pTDP-43和Ub的表达情况,结果显示,46.7%(14/30)的ALS患者三角肌、肱二头肌、肱三头肌的神经末梢中pTDP-43阳性表达,60.0%(18/30)的ALS患者Ub阳性表达。本研究中的pTDP-43阳性表达率与以往研究[6]报道的阳性表达率(33.3%)相比稍高,可能与入组病例数较少、检测肌肉部位不一致以及抗体特异性和灵敏度不一有关。此外,活检采取受试者的少量肌肉组织样本,取材的范围非常有限。
既往研究[4-5]亦证实,ALS转基因模型鼠的骨骼肌内具有多重病理表现。Atkin等[4]在ALS转基因模型鼠的腓肠肌、比目鱼肌和指长屈肌检测到可溶性超氧化物歧化酶-1蛋白聚集体。Wong等[5]发现只在骨骼肌中表达人类SOD1-G37R和SOD1-G93A的转基因小鼠,其神经组织中不表达上述突变蛋白,最终亦可发展成为ALS,致使其运动神经元受累、形成Ub阳性包涵体。上述研究为骨骼肌参与ALS发病这一理论提供了更为直接的证据。
ALS患者骨骼肌神经末梢中pTDP-43包涵体的产生及形成可能有多种机制。首先,可能是由于ALS患者肌细胞及神经纤维自噬-溶酶体系统和UPS损伤,导致TDP-43降解异常。自噬-溶酶体系统和UPS是真核细胞蛋白降解的重要途径。既往研究[14]表明,TDP-43降解途径包括自噬-溶酶体系统和UPS。P62是一种多功能Ub结合蛋白,参与自噬-溶酶体系统和UPS两种蛋白降解过程。Mizuno等[14]在28例ALS患者中观察到27例前角细胞中具有P62阳性包涵体。王建宇[15]通过对ALS患者骨骼肌标本行透射电镜观察,发现萎缩肌纤维内肌节结构紊乱,肌膜下溶酶体、线粒体聚集,可见次级溶酶体和自噬小泡。研究[16]发现ALS转基因小鼠骨骼肌组织自噬相关标记物表达上调。分析自噬-溶酶体系统可能参与了ALS骨骼肌病变过程,但其作用机制仍待进一步研究。Gilchrist等[17]将野生型Ub(H6Ub)和突变型Ub(H6UbK48R)分别转入ALS小鼠中,发现表达H6UbK48R的转基因小鼠的发病延迟,说明Ub的改变可影响ALS的进展。因此,自噬-溶酶体系统和UPS的功能异常可能参与了ALS 骨骼肌病变过程。另一可能机制是TDP-43在细胞间朊蛋白样播散,即通过顺轴突转运。有学者[18]用神经元-神经元跨突触播散理论来解释pTDP-43在脑和脊髓中的病理学播散,推测pTDP-43沿着运动神经元的轴突播散至神经肌肉接头。
综上所述,该研究结果提示除运动神经元、非运动神经元和神经胶质细胞以外,骨骼肌神经末梢可能是部分ALS患者pTDP-43病理沉积的又一部位。然而目前我国相关研究较少,而且也缺乏较为完整的流行病学资料,因此还需要更大样本量、部位更广泛的骨骼肌病理学研究去进一步证实。
