(Cu,N)共掺杂TiO2/MoS2异质结的电子和光学性能:杂化泛函HSE06∗
2018-12-14王冠仕林彦明赵亚丽姜振益张晓东
王冠仕 林彦明 赵亚丽 姜振益 张晓东
(西北大学现代物理研究所,陕西省理论物理前沿重点实验室,西安 710069)
(2018年8月12日收到;2018年9月29日收到修改稿)
1 引 言
在过去的几十年中,研究者已经尝试了大量的策略来解决以上问题.其中,常用方法是在光催化材料中掺杂各种杂质原子来调节材料的带隙,例如,非金属C,N和B掺杂TiO2[16−18],金属Fe和Cu掺杂TiO2[19,20],能够将光吸收边缘扩展到可见光范围并提高光催化效率.然而,单原子掺杂依旧存在一些缺陷,例如热稳定性低、电子-空穴复合高等.在此基础上,为了有效提高材料的光催化性能,对共掺杂体系进行研究,例如,非金属-非金属共掺杂、金属-金属共掺杂和金属-非金属共掺杂[21−23].此外,构建半导体异质结构是一种通过分离与转移光生电子和空穴来提高半导体材料的光催化活性的有效方法.例如,研究发现TiO2/g-C3N4异质结构能够有效分离光生电子和空穴,从而增强光催化产氢性能[24],SrTiO3/TiO2异质结有利于光生电子和空穴的快速分离,实现光催化性能的提高.因此,选择合适的共掺杂杂质原子以及构建合适的半导体异质结能够抑制光生电子-空穴对的复合和增强光催化活性.
最近,Jaiswal等[25]成功制备了(Cu,N)共掺杂TiO2材料,实验结果表明,(Cu,N)共掺杂TiO2可以增强对可见光的吸收同时降低光生电子空穴的复合.在光催化制氢的研究中,人们发现材料表面与水接触具有良好的光催化能力,而在自然界中,锐钛矿型TiO2的晶面暴露在真空外的仅有(101)和(001)两个表面,其中TiO2(101)表面占其中的94%,性能最稳定.现在有很多研究者对锐钛矿型TiO2(101)表面进行了理论研究,例如Sun等[26]和Yang等[27]研究的锐钛矿型TiO2(101)表面的光催化性.然而,据我们所知,关于(Cu,N)共掺杂的锐钛矿型TiO2(101)表面的电子结构与光学性质方面的理论研究很少.因此,本文通过理论计算分析共掺杂对TiO2(101)表面光吸收的影响.另外,已有研究表明构建TiO2/MoS2异质结可以有效抑制载流子复合,从而提高TiO2材料的光催化活性[28−33].因此,根据上述分析,可以预测(Cu,N)共掺杂的TiO2/MoS2异质结可以有效地分离和转移光生电子与空穴,同时增加对可见光的吸收.
在本文中,我们采用杂化泛函Heyd-Scuseria-Ernzerhof(HSE06)的方法系统地计算了Cu/N(共)掺杂的锐钛矿型TiO2(101)表面和Cu/N(共)掺杂的TiO2/MoS2异质结的几何结构、电子结构和光学性质.对于Cu/N(共)掺杂的锐钛矿型TiO2(101)表面和TiO2/MoS2异质结,分别计算了Cu/N原子掺杂在不同的间隙和替位掺杂位置的性质.另外,我们还分析了Cu/N(共)掺杂的TiO2/MoS2异质结界面处的电荷转移,研究了压力对TiO2/MoS2异质结电子结构和光吸收性质的影响.本文试图通过构建异质结、对异质结进行掺杂和加压来提高TiO2材料的光催化活性.
2 计算模型和方法
本文构建的基础模型有TiO2(101)表面和单层MoS2.为保证构建模型的稳定性,我们对其真空层进行测试,测试结果如图1所示.
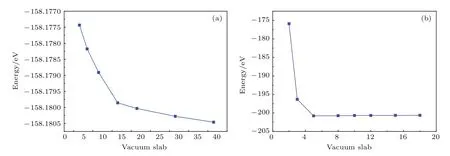
图1 不同真空层的(a)TiO2(101)表面和(b)单层MoS2的总能量Fig.1 .Calculated total energy for(a)TiO2(101)surface and(b)MoS2monolayer with dif f erent vacuum spaces.

图2 优化后的(a)锐钛矿型TiO2(101)表面,(b)单层MoS2和(c)TiO2/MoS2异质结Fig.2 .Optimized structures:(a)TiO2(101)surface;(b)MoS2monolayer;(c)TiO2/MoS2heterojunction.
如图1所示,TiO2(101)表面体系的总能量在真空层大于15 Å之后趋于稳定,因此本文在构建TiO2(101)表面时设置真空层为15 Å;单层MoS2体系的总能量当真空层大于6 Å后趋于稳定,因此单层MoS2的真空层被设为6 Å.锐钛矿型TiO2(101)表面包含24个氧原子和12个钛原子(共36个原子),优化后的结构如图2(a)所示.单层MoS2包含8个硫原子和4个钼原子(共12个原子),结构优化后如图2(b)所示.利用优化好的TiO2(101)表面和单层MoS2结构构建的TiO2/MoS2异质结如图2(c)所示.为得到稳定的TiO2/MoS2异质结,采用的真空层为15 Å,整个超胞的高度为35 Å.在研究掺杂对体系的影响时,考虑了Cu/N(共)掺杂TiO2(101)表面和TiO2/MoS2异质结的各种掺杂位置,例如N替位O(N@O)或N替位Ti(N@Ti),Cu替位O(Cu@O)或Cu替位Ti(Cu@Ti)等.
本文所有计算都使用VASP(Vienna ab initio simulation package)软件包采用PAW 方法进行[34],利用广义梯度中的Perdew-Burke-Ernzerhof(PBE)赝势来描述其交换关联和相关势[35].其截断能设置为400 eV,TiO2(101)表面和TiO2/MoS2异质结的几何优化和电子性质计算中k点网格分别采用5×5×1和3×3×1.优化结构设置能量和力的精度分别为10−5eV和0.01 eV/Å,为了获得更为准确的优化结果,采用了DFT-D2方法对范德瓦耳斯(van der Waals,VDW)相互作用进行理论修正,并用优化好的结构计算其电子和光学性质.为了准确计算材料的电子和光学性质,采用杂化泛函(HSE06)方法,交换关联能的表达公式如下:
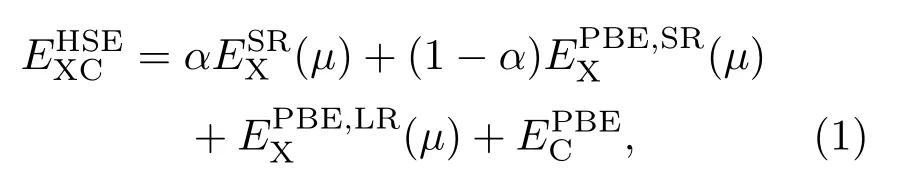
式中下标X和C分别表示交换泛函和关联泛函;µ为筛选参数,设置为0.20 Å−1;电子-电子相互作用可分为短程(shot range,SR)和长程(long range,LR)两部分,通过混合参数α来确定,α的默认设置为0.20,但不同材料α值不同.为了准确计算出材料的电子和光学性质,通过计算材料带隙测试了参数α的取值,结果如图3所示.由图3可以发现,α分别取0.05和0.17时单层MoS2和TiO2(101)表面的带隙与实验值(1.90 eV,2.90 eV)符合,在测出单层MoS2和TiO2(101)表面α值的基础上,通过对两个结构α值做加权平均,计算出TiO2/MoS2异质结的α值为0.14.
汉麻籽油的皂化、甲酯化根据GB/T 17376-2008/ISO 5509:2000“动植物油脂 脂肪酸甲酯制备”[11],选用三氟化硼法,每个样品皂化、甲酯化3次。
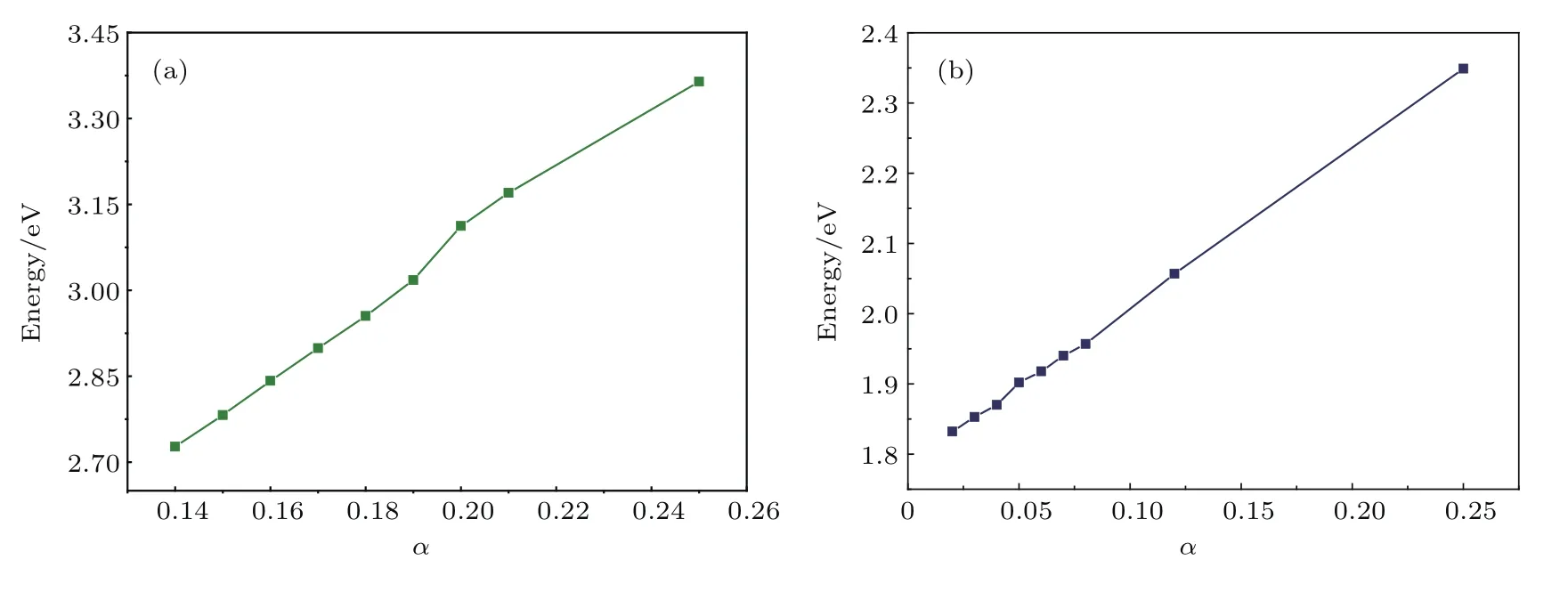
图3 α参数为不同值时(a)锐钛矿TiO2(101)表面和(b)单层MoS2的带隙Fig.3 .Calculated band gap with dif f erent α parameters:(a)TiO2(101)surface;(b)MoS2monolayer.
3 结果与讨论
3.1 几何结构
在研究掺杂体系和复合体系的性质前,首先分别研究了锐钛矿型TiO2(101)表面和单层MoS2结构的性质.钛矿型TiO2(101)表面优化后,晶格常数为a=b=3.786 Å,几何结构呈锯齿状,MoS2优化后的晶格常数a=b=3.19 Å,与以前的实验数据相符合[36−39],这表明我们的计算方法是正确的.
优化后的Cu/N(共)掺杂的TiO2(101)表面的几何结构如图4所示,相应体系的平均键长如表1所列.锐钛矿型TiO2(101)表面中Ti—O键的平均键长为2.001 Å,N掺杂TiO2(101)表面后,结构中Ti—O键的平均键长变化不明显,然而Cu掺杂对Ti—O键的影响相对较大,可能是由于Cu原子掺杂引起了局域晶格畸变.N原子掺杂体系中,Ti—N键的平均键长与Ti—O键的平均键长近似相等,这可能是由于N原子的半径(0.75 Å)与O原子(0.65 Å)的半径相差较小.由于Cu原子的半径(1.57 Å)远远大于O原子的半径(0.65 Å),所以Cu掺杂体系和(Cu,N)共掺杂体系中Cu—Ti的平均键长(2.479 Å,2.627 Å)远远大于纯的TiO2(101)表面中Ti—O的平均键长.这些结果表明,(Cu,N)共掺杂可以引起锐钛矿型TiO2(101)表面晶格发生明显的畸变,进而改变偶极矩,使得光生电子和空穴对更容易分离.在单层MoS2中,Mo—S的键长为2.411 Å,整个结构呈现为三明治形状,与实验结果相符合[40].
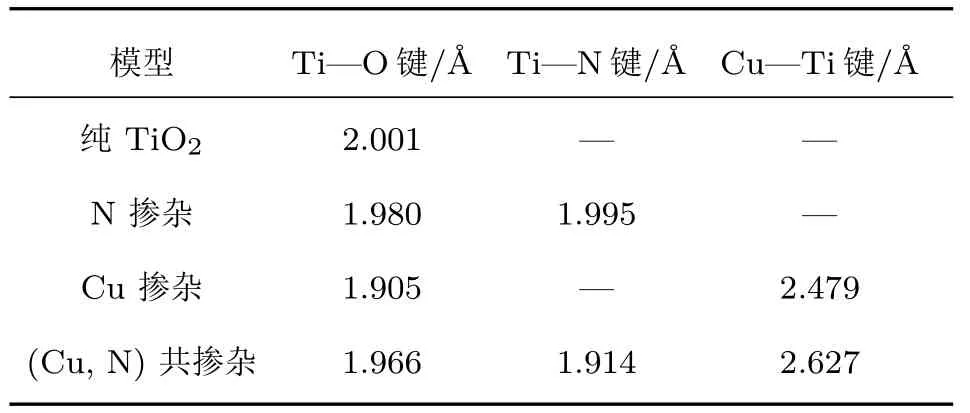
表1 优化后的Cu/N(共)掺杂的锐钛矿型TiO2(101)表面的平均键长(单位:Å)Table 1 .Average bond lengths of Cu or/and N doped anatase TiO2(101)surface after geometry optimization(unit:Å).
把优化好的Cu/N(共)掺杂TiO2(101)表面和单层MoS2构建出TiO2/MoS2异质结,再次对TiO2/MoS2异质结进行几何优化,其单层MoS2的厚度(h)和Cu/N(共)掺杂TiO2(101)表面与单层MoS2之间的垂直距离(D)的结果如表2所列.在纯的TiO2/MoS2异质结中,单层MoS2的厚度和TiO2(101)表面与单层MoS2之间的垂直距离分别为h=2.964 Å和D=2.891 Å.当掺杂N原子后,MoS2厚度(h)和TiO2(101)表面与单层MoS2之间垂直距离(D)变化不大.Cu掺杂和(Cu,N)共掺杂TiO2/MoS2异质结中单层MoS2的厚度(h)均有所增大,同时Cu掺杂和(Cu,N)共掺杂体系的TiO2(101)表面与单层MoS2之间的垂直距离(D)相比于纯的TiO2/MoS2异质结明显减小,这是由于较长的Cu—Ti键减小了Cu原子与单层MoS2之间的垂直距离,从而增强了TiO2(101)表面与单层MoS2之间的相互作用力.这些异质结中的垂直距离(D)均为范德瓦耳斯平衡间距.
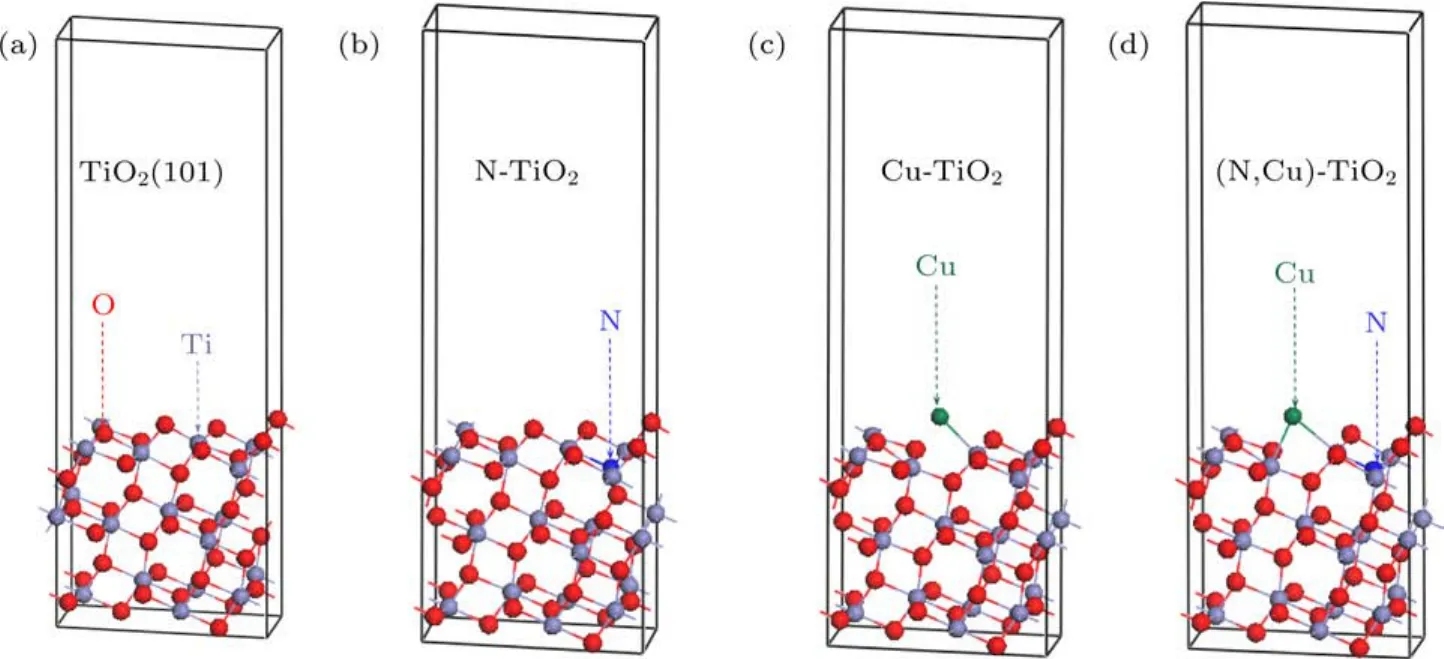
图4 优化后的TiO2(101)表面的超胞模型 (a)纯TiO2;(b)N掺杂TiO2(N@O);(c)Cu掺杂TiO2(Cu@O);(d)(Cu,N)共掺杂TiO2(Cu@O&N@O)Fig.4 .Optimized supercell models of anatase TiO2(101)surface:(a)Pure TiO2;(b)N doped TiO2(N@O);(c)Cu doped TiO2(Cu@O);(d)(Cu,N)codoped TiO2(Cu@O&N@O).
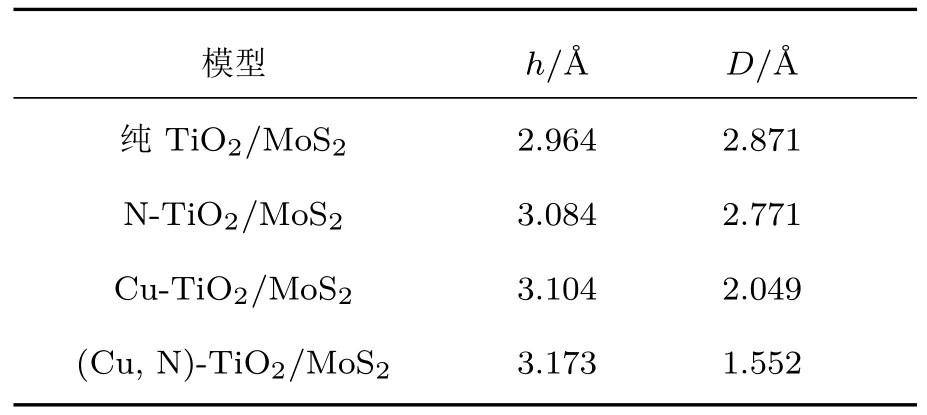
表2 单层MoS2的厚度(h)和单层MoS2与Cu/N(共)掺杂TiO2(101)表面之间的垂直距离(D)Table 2 .Thickness(h)of the MoS2and the vertical separation(D)between MoS2and Cu or/and N doped TiO2(101)surface.
3.2 缺陷形成能
为了解不同生长环境下掺杂体系的稳定性,我们根据以下公式计算了不同替位掺杂体系的形成能:
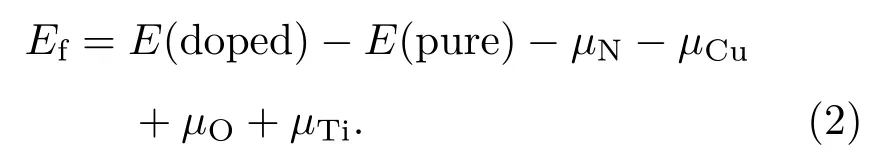
间隙位置掺杂体系的形成能计算公式如下:
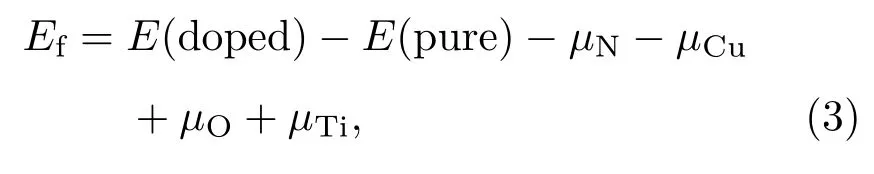
其中E(doped)和E(pure)分别表示掺杂体系和纯锐钛矿TiO2(101)表面的总能量.化学势µN和µO分别从自由分子N2(µN= µ(N2)/2)和O2(µO= µ(O2)/2)的基态能量中得出,Cu和Ti的化学势分别从其块状材料基态能量得出.然而,形成能计算有富Ti和富O两种情况[41].结构中Ti和O的化学势满足µTi+2µO= µ(TiO2),在富Ti条件下,µTi取块体Ti的化学势,O的化学势为µO=(µ(TiO2)− µTi)/2. 在富O条件下,µO取O2分子的基态能量(µO= µ(O2)/2),µTi= µ(TiO2)− µ(O2).
不同掺杂情况下缺陷形成能计算结果如表3所示,Ef越小就意味着杂质原子越容易在TiO2(101)表面掺杂.在选择替位掺杂位置时,计算结果显示替换TiO2(101)表面顶层原子(靠近真空层位置)的形成能相比于替位内部原子(远离真空层位置)都稍小一些,因此,替位顶层原子的结构更容易形成.而TiO2(101)表面的顶层存在配位数分别为2和3的两种O原子,通过分别计算其缺陷形成能得出Cu和N替位配位数分别为2和3时的O原子时形成能最小.因此,下文中的Cu@O,N@O体系指所替位的O原子的配位数分别为2和3的顶层氧原子的掺杂体系.对于Cu/N(共)掺杂的TiO2(101)表面,在富Ti生长条件下,Cu@O和N@O的缺陷形成能远小于其他掺杂体系,其几何形状如图4(b)和图4(c)所示.在富O的生长环境下,Cu@Ti的缺陷形成能远小于其他掺杂体系.结果表明,在富Ti条件下,O离子更容易被N和Cu杂质取代,而在富O条件下,Ti离子更容易被Cu杂质取代.对于Cu和N共掺杂的TiO2(101)表面(表3),在富Ti生长条件下,Cu@O和N@O的缺陷形成能远小于其他共掺杂位置的体系,几何结构如图4(d)所示.尤其在富Ti条件下,Cu@O和N@O的缺陷形成能为−9.146 eV,表明Cu@O和N@O的替位掺杂很容易形成.同样,对于Cu或/和N掺杂的TiO2/MoS2异质结,在富Ti条件下,Cu@O和N@O的缺陷形成能为最小,表明Cu@O和N@O很容易在异质结中形成.因此本文选取Cu和N替代O的结构来计算结构的相关性质.
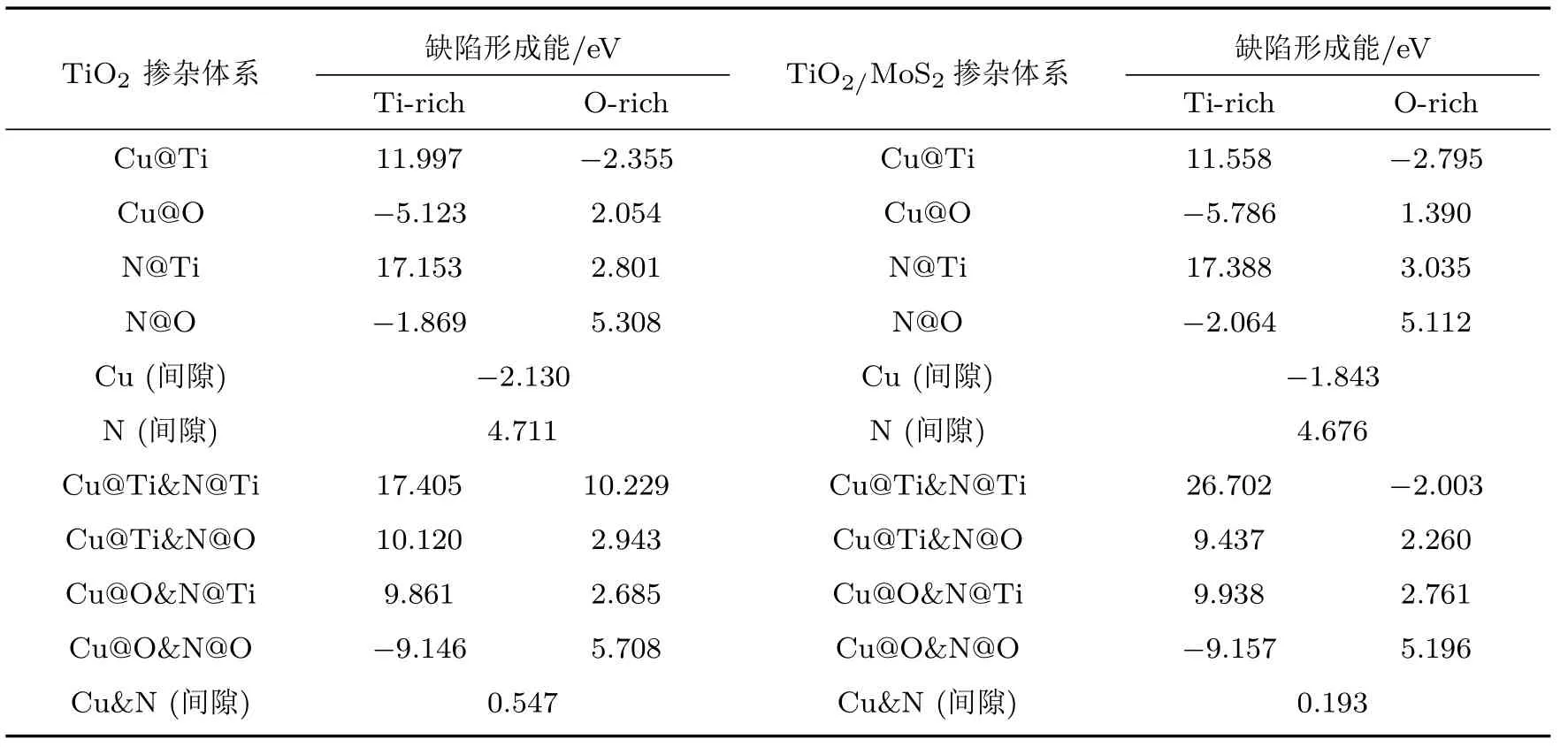
表3 Cu/N(共)掺杂的锐钛矿TiO2(101)表面和Cu/N(共)掺杂的TiO2/MoS2异质结的缺陷形成能Table 3 .Defect formation energy of Cu or/and N doped anatase TiO2(101)surface and TiO2/MoS2 heterostructure.
3.3 电子态密度和电荷转移分析
为了研究Cu/N(共)掺杂对TiO2/MoS2异质结光催化活性和光吸收性能的影响,通过杂化泛函计算了Cu/N(共)掺杂的TiO2(101)表面和Cu/N(共)掺杂TiO2/MoS2异质结的态密度,结果如图5所示.
图5描述了所有体系的总态密度(total density of states,TDOS)和对应的所有体系分波态密度(partial density of states,PDOS).由图5(a)和图5(a′)可以发现,TiO2(101)表面的带隙为2.90 eV,其价带顶(value based management,VBM)主要由O 2p态提供,导带底(conduction band minimum,CBM)主要由Ti 3d态组成,这与实验结果相符合,表明本文计算结果的正确性.与纯的TiO2(101)表面态密度相比,N掺杂后对TiO2(101)表面的导带和价带位置影响不大,但在带隙中存在N 2p的杂质态(图5(b)和图5(b′)),这种杂质态可以增强材料在可见光范围的光吸收,提高材料对可见光的利用.Cu掺杂体系的禁带中存在Cu 3d杂质态(图5(c)和图5(c′)),同样能够提高材料的光催化活性.由图5(d)和图5(d′)得出在(Cu,N)共掺杂的TiO2(101)表面中,由于Cu和N的协同作用,使得体系的带隙明显减小,这样能有效提高材料对可见光的利用率.

图5 (共)掺杂体系的TDOS和PDOS图 (a)和(a′)纯的TiO2(101)表面;(b)和(b′)N掺杂的TiO2(101)表面;(c)和(c′)Cu掺杂的TiO2(101)表面;(d) 和(d′)(N,Cu)共掺杂TiO2(101)表面;(e)和(e′)纯的TiO2/MoS2;(f)和(f′)N-TiO2/MoS2;(g)和 (g′)Cu-TiO2/MoS2;(h)和 (h′)(N,Cu)-TiO2/MoS2(费米能级设置为 0)Fig.5 .Calculated TDOS and PDOS of(co)doping systems:(a)and(a′)Pure TiO2(101)surface;(b)and(b′)N doped TiO2(101)surface;(c)and(c′)Cu doped TiO2(101)surface;(d)and(d′)(N,Cu)doped TiO2(101)surface;(e)and(e′)pure TiO2/MoS2;(f)and(f′)N-TiO2/MoS2;(g)and(g′)Cu-TiO2/MoS2;(h)and(h′)(N,Cu)-TiO2/MoS2(The Fermi level is set to zero).
关于TiO2/MoS2异质结的态密度,计算结果表明TiO2/MoS2异质结的带隙(1.38 eV,见图5(e))比纯的TiO2(101)表面带隙(2.90 eV)明显减小.这意味着在TiO2/MoS2异质结中电子能容易地从价带转移到导带,导致光吸收能力提高.在Cu和N掺杂TiO2/MoS2异质结后,态密度的结果如图5(f)和图5(g)所示,体系的带隙有所减小,但变化不明显.当(Cu,N)共掺杂TiO2/MoS2异质结时(图5(h)),由于Cu和N协同作用,在带隙中费米面附近出现了Cu 3d轨道提供的杂质态,这种杂质态可以起到桥梁作用,使低能光生电子从价带先跃迁到杂质带再到导带,从而增强对低能光子的吸收,可以促进电子空穴对分离,提高材料对可见光的利用率.
为了更清晰地了解Cu/N(共)掺杂异质结体系中TiO2(101)表面和MoS2单层之间的电荷转移,计算了Cu/N(共)掺杂TiO2/MoS2异质结的差分电荷密度.计算三维差分电荷密度的公式如下:
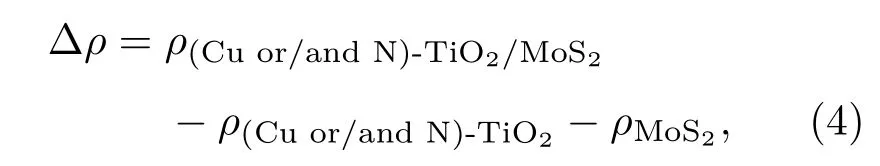
其中ρ(Cu or/and N)-TiO2/MoS2,ρ(Cu or/and N)-TiO2和ρMoS2分别表示Cu/N(共)掺杂的TiO2/MoS2异质结、Cu/N(共)掺杂的TiO2(101)表面和单层MoS2的电荷密度.除此之外,通过沿x-y平面的积分电子密度差法计算了平面平均差分电荷密度,公式如下:

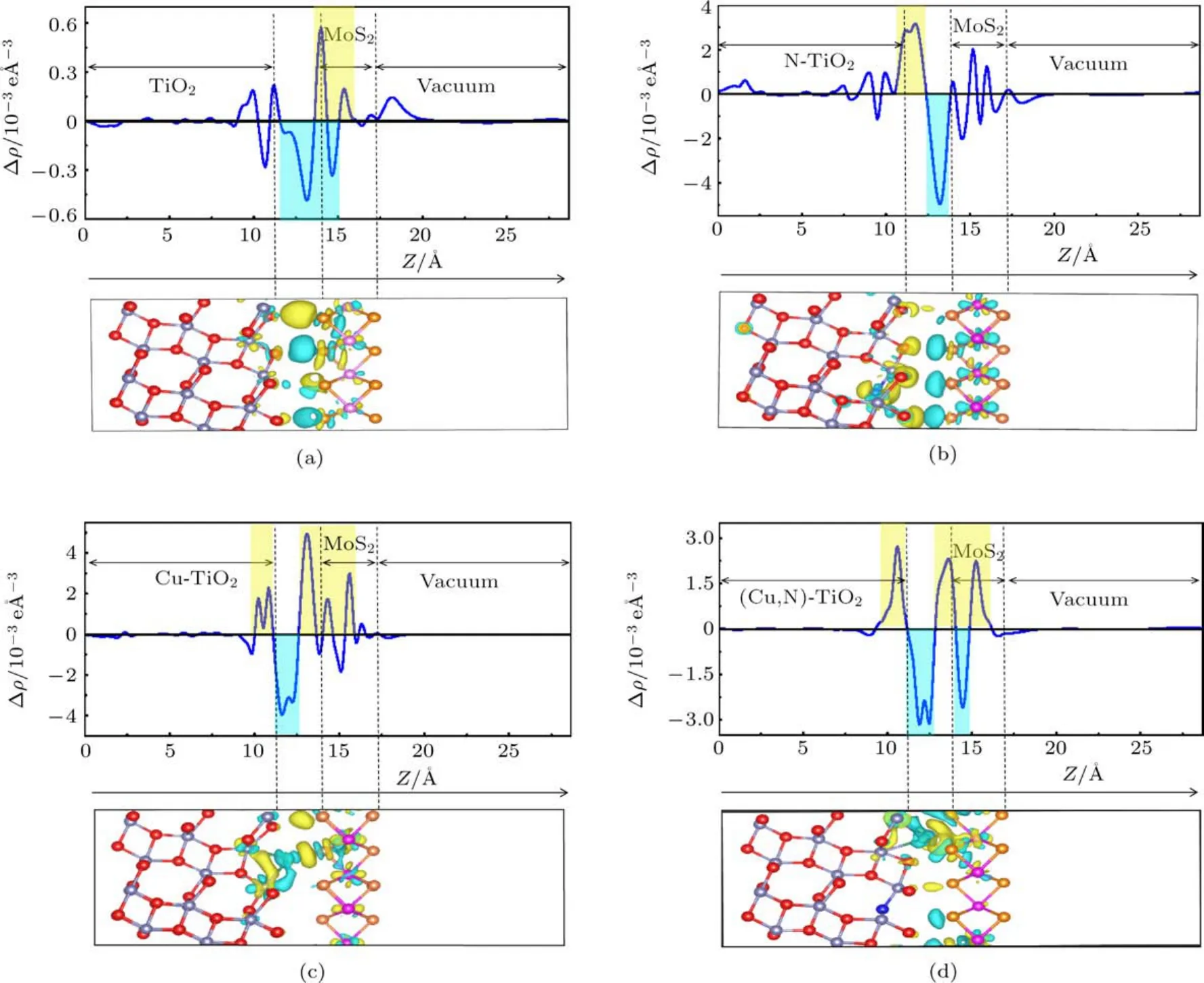
图6 (a)纯TiO2/MoS2,(b)N-TiO2/MoS2,(c)Cu-TiO2/MoS2,(d)(Cu,N)-TiO2/MoS2的差分电荷密度图(每个分图上面的是z方向上的平面平均差分电荷密度,正值表示电子积累,负值表示电子耗尽;下面是三维差分电荷密度图,黄色和青色区域分别表示电子积累和耗尽)Fig.6 .Charge density dif f erence:(a)Pure TiO2/MoS2;(b)N-TiO2/MoS2;(c)Cu-TiO2/MoS2;(d)(Cu,N)-TiO2/MoS2(Top of a function of position in the z-direction,the positive value indicates electron accumulation,and the negative value indicates electron depletion.Bottom of three dimensional charge density dif f erence,the yellow and cyan areas indicate electron accumulation and depletion,respectively).
其中z轴是沿界面法线的方向,x-y平面是垂直于z轴的超截面;i和j分别表示a轴和b轴格.计算结果如图6所示.
图6中分别描述了TiO2/MoS2体系、 NTiO2/MoS2体系、Cu-TiO2/MoS2体系和(Cu,N)-TiO2/MoS2体系平面和三维差分电荷密度图.在平面差分电荷密度图中,正值方向表示电荷积累,负值方向表示电荷耗尽.在三维差分电荷密度图中,青色区域表示电荷耗尽,黄色区域表示电荷累积.三维差分电荷密度图表明,在Cu/N(共)掺杂的TiO2(101)表面和MoS2单层之间电荷发生转移.图6(a)TiO2/MoS2体系差分电荷密度图可以看出,电荷由TiO2(101)表面转移向MoS2单层.由图6(b)可以看出,当N原子掺杂TiO2/MoS2异质结后,电荷由MoS2转移向TiO2(101)表面,并且电荷转移量也有所增加,这是N原子掺杂引起了电荷转移的变化.在Cu掺杂和(Cu,N)共掺杂的TiO2/MoS2异质结中,电荷由异质结界面附近的原子分别转移向TiO2和MoS2两侧(图6(c)和图6(d)),这可能是由于Cu原子附近的电荷发生转移引起的.以上结果表明,本文研究的几种掺杂能够有效提高复合体系电荷转移能力,抑制光生电子和空穴的复合,从而提高材料的光催化活性.
3.4 光学性质
作为光催化剂,高效吸收太阳光是它的基本需求,尤其是对可见光的吸收.为了探索对可见光吸收的机理,通过理论计算了材料对紫外和可见光吸收光谱.为了很好地比较,通过偶极子近似中的费米黄金法则,计算了纯TiO2(101)表面、单层MoS2,Cu/N(共)掺杂的TiO2(101)表面和Cu/N(共)掺杂的TiO2/MoS2异质结的频率相关介质矩阵.介电函数的虚部表达式如下:
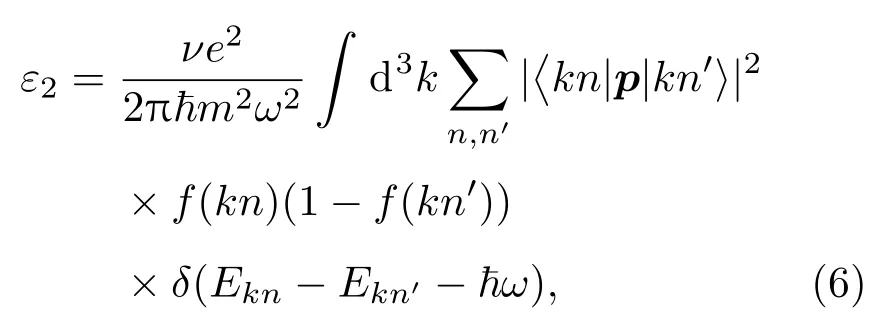
其中 ~ω是入射光子的能量,代表动量算符;r(h/i)(∂/∂x)是电子波函数;f(kn)是费米函数.通过Kramer-Kronig变换从虚部求出介电函数的实部. 吸收系数I(ω)由ε1(ω)和ε2(ω)导出, 公式如下:

考虑到介电函数的张量性质,ε1和ε2取沿x,y,z方向的平均值.而在计算吸收系数时,按照目前VASP程序的理论,考虑了带间跃迁,例如在(6)式中,计算虚部时涉及到带间跃迁的kn和kn′两个态.上述关系是能带结构和光学性质分析的理论基础,反映了不同能级之间电子跃迁引起的吸收光谱机理.
半导体光催化剂的光学性质是其光催化性能的关键因素之一. 因此,本文详细计算了TiO2(101)表面和Cu/N(共)掺杂的TiO2(101)表面的吸收光谱,结果如图7所示.图7(a)描述了Cu/N(共)掺杂的TiO2(101)表面的光吸收图,并将计算结果与实验吸收光谱[42](图7(b))进行了比较.图7(c)中包括了TiO2/MoS2异质结、Cu/N(共)掺杂的TiO2/MoS2异质结的吸收光谱.单层MoS2,TiO2(101)表面和TiO2/MoS2异质结的吸收光谱如图7(d)所示.
结果表明,由于锐钛矿型TiO2(101)表面的禁带宽度(2.90 eV)较大,所以它只能吸收紫外光,而对可见光不能有效利用.通过给TiO2(101)表面掺杂能够有效改善这一缺陷.
例如N掺杂TiO2(101)表面后,体系的带隙变为2.58 eV,在近紫外光区域中和可见光(λ<500 nm)区域光吸收效率明显增加.对于Cu掺杂的TiO2(101)表面,Cu 3d轨道影响体系的禁带宽度,使体系在可见光区域内具有良好的光吸收率.对于(Cu,N)共掺杂TiO2(101)表面,Cu和N杂质共掺杂的协同效应导致禁带宽度缩小,使体系对可见光的吸收大大增强.结果可能是(Cu,N)共掺杂TiO2(101)表面导致可见光光催化活性提高和光吸收边缘明显红移.计算结果与实验数据相符合(图7(b)).为了提高TiO2(101)表面的光催化活性,构建出TiO2/MoS2异质结.计算TiO2/MoS2异质结的光吸收性能,结果如图7(d)所示.从图7(d)可以看出,TiO2/MoS2异质结相比TiO2(101)表面的可见光来说,其光催化活性提高了,光吸收边缘明显发生了红移.结果表明,构建异质结能够有效提高材料对太阳光的利用.为了进一步提高材料对太阳光的利用,我们构造Cu/N(共)掺杂到TiO2/MoS2异质结中TiO2(101)表面的结构,计算出各种结构的光吸收性能,结果如图7(b)所示.与TiO2/MoS2异质结的光学性质相比,由于N 2p轨道影响,N-TiO2/MoS2体系在近紫外光区域和可见光(λ<630 nm)区域中的光吸收性能明显增加.计算Cu-TiO2/MoS2体系的光催化性能,结果表明它在可见光范围内的光吸收略有增强.相比于单掺杂体系,(Cu,N)共掺杂TiO2/MoS2体系对于太阳光的吸收能力有着显著的提高.总而言之,异质结的形成和Cu/N(共)掺杂到TiO2/MoS2异质结都可以增强光吸收,并改变吸收边缘.
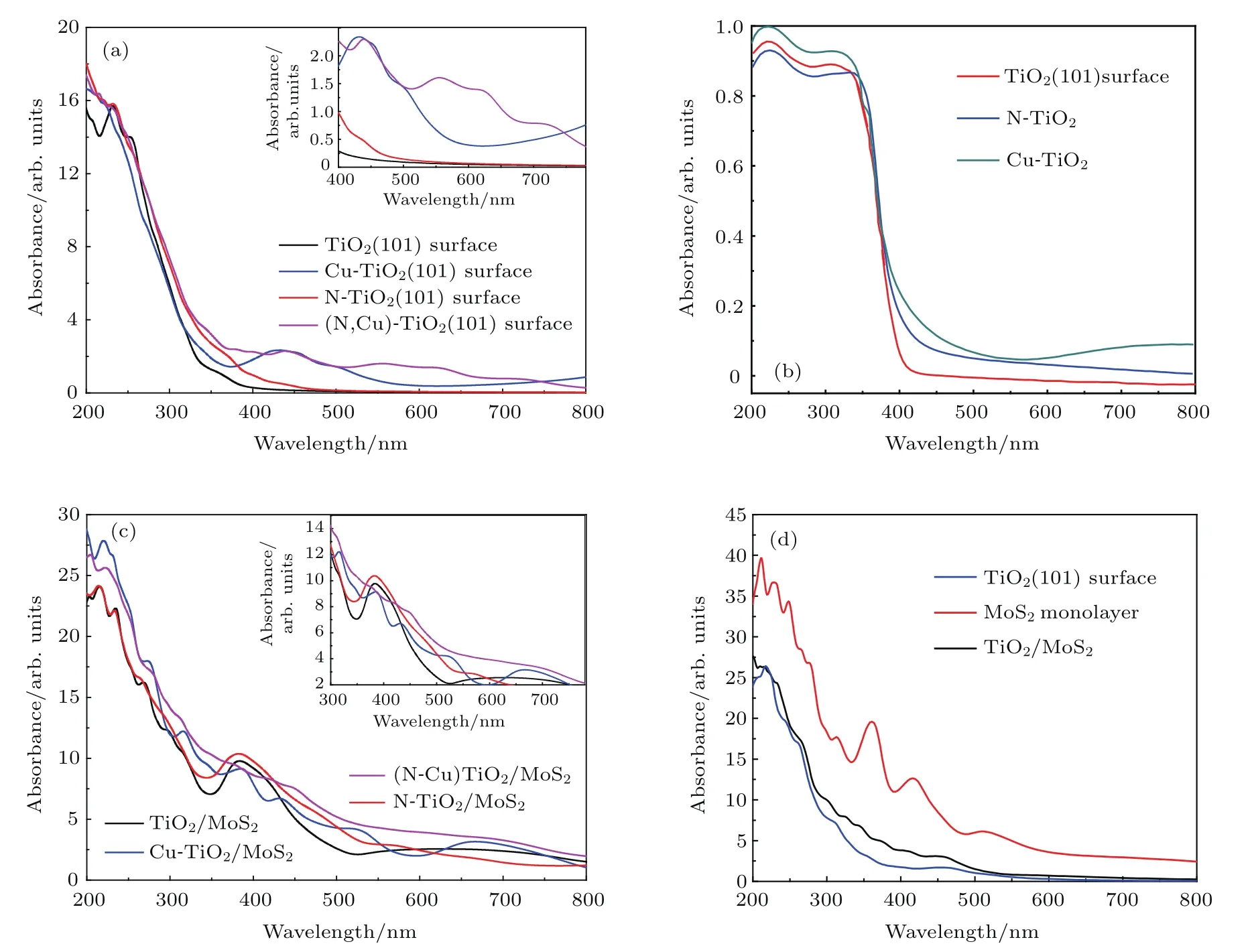
图7 (a)掺杂TiO2(101)表面的光学吸收光谱;(b)掺杂TiO2(101)表面的光学吸收光谱的实验数据;(c)掺杂TiO2/MoS2异质结的光学吸收光谱;(d)TiO2(101)表面、单层MoS2和TiO2/MoS2异质结的光学吸收光谱Fig.7 .(a)Optical absorption spectra of doping TiO2(101)surface;(b)experimental data of optical absorption spectra of doping TiO2(101)surface;(c)optical absorption spectra of doping TiO2/MoS2heterostructure systems;(d)optical absorption spectra of TiO2(101)surface,MoS2monolayer and TiO2/MoS2heterostructure systems.
3.5 压力对TiO2/MoS2异质结的影响
在以上研究的基础上,继续研究了压强对TiO2/MoS2异质结复合材料光催化性能的影响.采用的模型为优化好的TiO2/MoS2异质结.对TiO2/MoS2异质结分别施加10,20,30,40,50 GPa的静水压,并计算在不同压力下的电子结构和光学性质.结果发现,压强增加导致体系的晶格常数以及体系的真空层都逐渐减小,整个结构均处于压缩状态.
图8(a)中给出了体系总能量随压强变化的关系,从图8(a)中可以看出,从10 GPa到50 GPa体系总能随压强线性增长,这说明体系随着压强的增加稳定性在减弱.因为在TiO2/MoS2异质结几何结构优化后,晶体结构中的各原子都达到了最稳定的平衡状态,总能量达到最低状态,但随着外界压强的增大,晶体势能逐渐增大,从而体系的总能量也增加.图8(b)和图8(c)分别描述了体系的态密度和光学性质.从图8(b)中可以发现,随着压强的增加体系禁带宽度逐渐减小,但不同范围的压强对带隙影响大小不一样.例如,在体系分别加0 GPa和10 GPa的压强时,两状态体系的禁带宽度无明显变化,说明体系加10 GPa的压强对体系电子性能影响不大.当体系施加的压强逐渐从10 GPa增加到30 GPa时,体系带隙明显减小(0.15 eV).然而当给体系施加压强增加到40 GPa时,体系禁带宽度相对于30 GPa出现明显减小(0.35 eV),这可能是由于体系发生相变导致.当压强增加到50 GPa时,体系的带隙变为0.30 eV.这表明,通过加压可以有效地减小体系的带隙.从图8(c)中可以看出,当给体系施加10 GPa的压强后,复合材料对近紫外区的吸收能力有显著的提高,并对400—700 nm范围的可见光吸收能力也有提高,吸收波峰向可见光转移.随着对复合材料继续加压,复合体系对近紫外和可见光的吸收性能依次提高.当对复合体系的压强施加到50 GPa时,复合体系对450—800 nm区域的光吸收能力有明显提高.这表明,通过给复合材料加压可以有效提高材料的光学性质.

图8 TiO2/MoS2异质结在不同压强下的(a)结合能,(b)光吸收谱和(c)态密度Fig.8 .(a)Formation energy diagram,(b)optical absorption map and(c)density of state of TiO2/MoS2 heterostructure at dif f erent pressures.
4 结 论
本文采用杂化密度泛函方法对TiO2/MoS2异质结、Cu/N(共)掺杂的TiO2(101)表面和Cu/N(共)掺杂的TiO2/MoS2异质结结构的电子结构和光学性质进行了研究.结果表明,TiO2/MoS2异质结比纯TiO2(101)表面的带隙明显减小,(Cu,N)共掺杂诱导TiO2/MoS2异质结带隙中出现了N 2p和Cu 3d杂质态,减小了光生电子-空穴的复合率,提高了材料对可见光的吸收能力.计算了Cu/N(共)掺杂的TiO2/MoS2异质结的差分电荷密度,发现电荷最终会由异质结的一侧转移到另一侧,有效抑制了光生电子-空穴的复合.因此,Cu/N(共)掺杂的TiO2/MoS2异质结光催化剂具有令人满意的光催化活性.除此之外,计算了压力对复合材料光催化性能的影响,发现通过施加压力可以有效提高材料的光催化活性.这些理论计算可以为实验提供一个引导与理论解释.
