锂离子电池LiMn2O4和LiNi0.5Mn1.5O4正极材料的电子结构
2018-11-01朱彦荣伊廷锋岳彩波
桂 轩,朱彦荣,谢 颖,伊廷锋,岳彩波
(1.安徽工业大学化学与化工学院,安徽马鞍山243002;2.黑龙江大学 功能无机材料化学教育部重点实验室,黑龙江哈尔滨150080)
LiMn2O4系列材料以其原料资源丰富、成本低廉(Mn与Co的价格比为1/40~1/20)、安全性好、无环境污染、制备容易等优点,一度成为动力锂离子电池正极材料的希望,但由于在充放电过程中会发生Jahn-Teller效应,导致温度高于55℃时,材料结构发生变形,且晶体中的Mn3+会发生歧化反应,生成的Mn2+溶解于电解质中使电极活性物质损失,容量衰减很快,这些均阻碍和限制了LiMn2O4进一步的研究、开发和应用[1]。通过优化反应条件及改进合成方法等途径来改善尖晶石LiMn2O4正极材料的性能取得了一定成效,但并不能从根本上解决LiMn2O4多次循环后的容量损失问题,且单独开展提高其电化学性能的工作有一定局限性。LiMn2O4属于Fd3m空间群,锰离子(3d3,3d4)占据八面体16d位,很容易被锂离子(2s0)取代,形成非化学计量比锂锰氧化物,而不引起结构变化。因此,采用其他离子对锰离子进行掺杂,可以充分抑制Jahn-Teller效应,有效提高电极的循环寿命,抑制容量的衰减[2]。近年来,随着耐高电压电解液的研制成功,采用过渡金属离子对锰离子进行掺杂,生成尖晶石相LiM0.5Mn1.5O4(M=Cr、Ni、Cu、Fe),可以将电池的充放电电压提高到5 V左右,充分抑制Jahn-Teller效应的发生,有效地提高电极的循环寿命,从而引起人们的广泛关注[3]。电池的容量和充放电平台电压取决于过渡金属离子的类型和浓度,5 V电池的优点在于可以获得高的功率密度[4]。过渡金属离子掺杂锰酸锂正极材料具有比较高的输出电压,充分抑制充放电过程中结构的不可逆变化,提高锂离子电池的循环性能、能量密度以及功率密度。大量研究表明,只有材料LiNi0.5Mn1.5O4表现出可接受的稳定性能[5],其首次放电容量在很多报道中均能达到140 mAh·g-1左右,接近理论容量,充放电时没有4.0 V平台,不存在Mn3+/Mn4+的氧化-还原过程,只在4.7 V处有一个充放电平台,对应Ni2+/Ni4+的氧化-还原过程,充放电50次后LiNi0.5Mn1.5O4的容量保持率在96%以上[6]。
目前锂离子电池电极材料的掺杂改性方案的设计仍存在一定的随机性,缺乏相应的理论依据。该问题的产生主要源于人们对其结构和性能关系的认识不够清晰。第一性原理计算方法的优势在于不论所研究的材料是否易于合成、材料归属于何种结构,皆能准确地计算出各物质的能量,进而计算出平均电势,为揭示材料的结构和电化学性质之间的关系提供理论依据。根据计算结果对已经合成材料的某些性能进行预测,并从理论上对某些试验结果加以解释,通过第一性原理计算方法可找出一些普遍性的规律,可为初步确立掺杂改性和材料的设计提供依据[7]。目前有关LiMn2O4和LiNi0.5Mn1.5O4材料的第一性原理计算已经取得了一些进展,如:杨思七等[8]计算表明少量的过渡金属M取代LiNi0.5Mn1.5O4晶格中的Ni,能够有效抑制材料在电化学脱嵌锂过程中的体积变化,提高材料循环性能。Shi等[9]计算Ni掺杂对LiMn2O4的结构以及电化学性能的影响,结果表明LiNi0.5Mn1.5O4具有高嵌入电压是由于新O2p带的出现所致。为了揭示异质元素的掺入对LiMn2O4材料的结构和性能的影响,本文拟采用第一性原理方法对LiMn2O4及其Ni掺杂的LiNi0.5Mn1.5O4材料的电子结构和化学键特征展开研究,其中能带结构、差分密度及电子密度的相关分析将为阐明这类材料的结构和性能关系奠定理论基础。
1 计算方法
计算基于CASTEP软件[10],采用平面波赝势技术,平面波的截断半径为380 eV。布里渊区采用(3×3×3)的网格面,几何优化采用BFGS算法[11]。测试表明采用GGA-PW泛函[12]计算所得到的晶格常数与实验吻合,误差小于3%。在计算过程中考虑到过渡金属的交换相关作用及自旋态问题,采用自旋极化和GGA+U的方法对系统的能带结构及电子性质进行处理[13]。Mn和Ni的库仑作用设置为5.0 eV。各离子步自洽场迭代的收敛标准为:在两次迭代过程中的能量差小于1.0×10-5eV/atom。计算过程中各原子的组态分别为:Li:1s22s1,O:2s22p4,Mn:3d54s2,Ni:3d84s2。LiMn2O4和 LiNi0.5Mn1.5O4的计算模型如图1所示。由于周期边界条件下采用晶胞和原胞所得到的结果是一致的,因此计算过程采用原胞,不仅可以有效地降低计算量,且原胞中4个Mn等价,以1个Ni取代任意1个Mn即可实现LiNi0.5Mn1.5O4的计量比。在尖晶石LiNi0.5Mn1.5O4中,Ni和Mn与氧形成八面体,过渡金属(Ni和Mn)均占据16d位,O占据32e位,Li位于晶格中的8a位。
2 结果与分析
通过计算材料的本征态在动量空间中的数值可以得到材料的能带结构图。LiMn2O4及其掺杂化合物在[-10 eV,10 eV]区间的能带分布如图2,3所示。能带结构图中的横坐标为动量空间,其与晶胞的对称点群有关,纵坐标为本征态的能量。
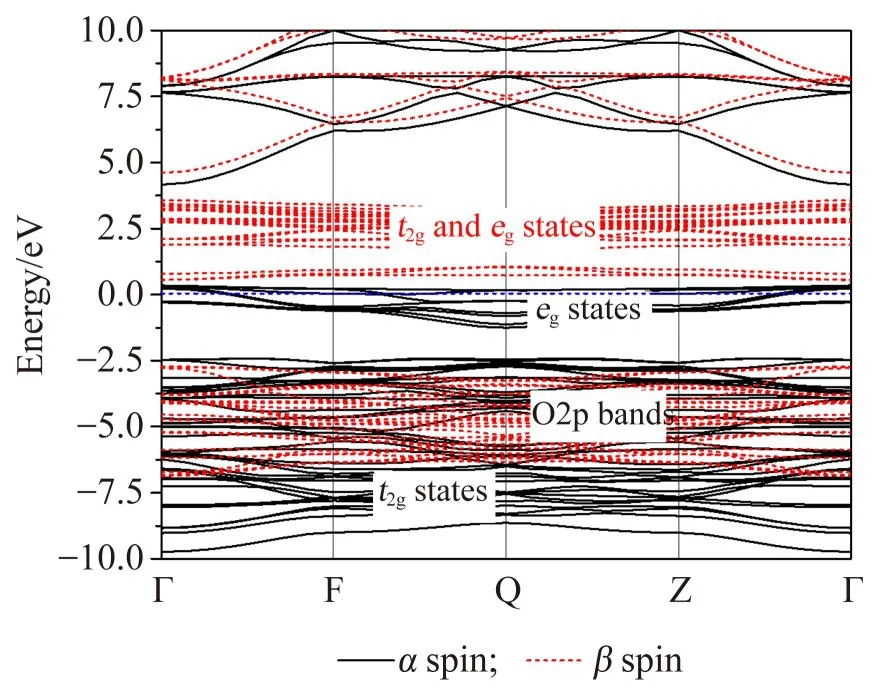
图2 LiMn2O4的能带结构Fig.2 Energy band structure of LiMn2O4

图3 LiNi0.5Mn1.5O4的能带结构Fig.3 Energy band structure of LiNi0.5Mn1.5O4
由图2,3可以看出,两种物质在-8~-2 eV区间的能级分布比较密集,该区间的能带主要是以O2p能带为主,同时有部分能带归属于Ni和Mn,这种现象与过渡金属的库仑排斥作用及电荷转移项有关,各能带之间的相对位置取决于两者之间的平衡[12-13]。由图2可知:LiMn2O4中Mn的库仑排斥项明显比电荷转移项的数值大,该材料的能带结构具有明显的电荷转移现象,即为非Mott-Hubbard材料[13];LiMn2O4材料的α通道的部分3d态将位于O2p能带之—;此外,处于[-2 eV,4 eV]区间的能带主要贡献来自于Ni和Mn,这是由于在NiO6和MnO6八面体中与O2p之间形成很强的σ键,导致它们的能量位于O2p能带之上;处于[4 eV,8 eV]区间附近且分裂较明显的两条能带归属于Li的2s态,因此Li2s轨道上的电子将转移到O2p态上,导致其被腾空;Li主要以离子的形式存在于LiMn2O4及其掺杂化合物的晶格中。
从能带结构的计算结果可以清楚地看出LiMn2O4及其掺杂化合物的α和β自旋态发生了明显的分裂,这表明过渡金属离子具有磁性。对于LiMn2O4而言,根据电荷平衡条件可,Mn的平均价态为3.5+。Mn原子的组态是3d54s2,属于d7结构,因此Mn3.5+离子的价电子构型介于d3和d4之间。根据能带结构(图2)的计算结果,对于LiMn2O4的β自旋通道,最高占据轨道(价带顶)主要来源于O2p带,最低非占据轨道(导带底)主要源于Mn的3d态,两者之间带隙约为3.3 eV,因此可以判断Mn离子在β自旋通道的3d态是非占据态,其自旋电子排布为t2g0eg0。同样在α自旋通道,LiMn2O4中的Mn的t2g态是完全填充态,而剩余的部分电子将填充在eg轨道上,这导致系统的费米能级(E-Ef=0 eV)经过eg轨道,Mn具有约3.5μb的平均磁矩且呈高自旋排布。
当Mn部分被Ni取代之后,系统的电子结构发生了明显的变化:O2p能带与部分Ni3d和Mn3d在[-7.5 eV,-2.0 eV]区间发生明显的混合;LiNi0.5Mn1.5O4中Mn的β自旋通道仍保持非占据状态,即t2g0eg0。由于Ni原子(3d84s2)的外层价电子较多,因此Ni离子在β自旋通道是满占据的,对应的自旋结构为t2g3eg2。与β自旋通道自旋通道相比,当Ni取代Mn以后材料的α自旋通道的能带结构也发生了相应的变化:Ni/Mn离子在费米能级之上的eg非占据态的数目明显少于它们在β自旋通道中的能级数目;Ni和Mn离子在该通道的自旋构型均为t2g3eg0。因此,在LiNi0.5Mn1.5O4材料中Mn和Ni离子的自旋构型分别为t2g3eg0和t2g6eg2,且它们的理论磁矩分别为3μb和2μb;Mn和Ni离子的价态分别为4+和2+。需要注意的是由于Ni部分替代Mn以后β自旋通道价带顶和导带底之间的带隙明显减小(约为1.2 eV),因此可以预期Ni的掺杂对于提高材料的导电性是有利的。此外,能带结构的计算结果也表明,5 eV之上的能带分别归属于Li的2s以及金属M的4s和4p能带。由于它们是非占据态,且与O2p的共价作用相对较弱,此处不再进行讨论。
为了进一步研究原子之间相互作用的本质,还计算了两个系统的差分电子密度,结果如图4,5。在差分电子密度图中,圆圈区域表示电子密度得到增强的区域,三角形区域表示电子密度减弱的区域。从计算结果可以看出,在LiMn2O4系统中,相邻的两个Mn离子的差分电子密度有较大的差异,表明Mn具有两种价态,即4+和3+,平均价态为3.5+,这与能带结构分析结果一致。此外,由于共价键的作用,系统的电子密度重新分布,其中:Mn离子附近电子密度减弱的区域呈对称的花瓣型,为典型的Mn3d轨道的特征;O原子在截面上的差分电子密度为三角形,表明O有sp3杂化,并且这些杂化轨道与三个过渡金属相连接,Mn3d轨道和O2p轨道之间具有很强的相互作用,形成了强共价键。另外Li与O之间的电荷密度较弱,可以推断Li和O之间是离子键作用,这与前面的计算结果一致。当Ni部分取代Mn以后,除了Mn和O之间的共价作用以外,Ni3d和O2p之间也形成了较强的相互作用。但由于Ni的部分eg态是部分占据的,因此可以推测Ni—O之间的相互作用较Mn—O之间的相互作用弱,这与能带结构的计算结果完全一致。此外,由于Ni的掺入,在锂离子嵌入和脱出过程中,Mn离子并不参与反应,其价态仍能够保持4+,在充放电过程中锰不会被还原成低价态,较好地解决了Mn的溶出问题,提高了材料的结构完整性;Ni在锂离子从LiNi0.5Mn1.5O4脱嵌后,其将被氧化成4+,从而成为该材料的氧化还原中心。

图4 LiMn2O4的三维差分密度(左)和二维电子密度(右)Fig.4 3D difference diagram(left)and 2D electron density(right)for LiMn2O4
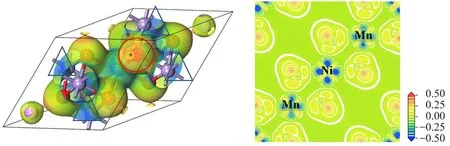
图5 LiNi0.5Mn1.5O4的3D差分密度(左)和2D电子密度(右)Fig.5 3D difference diagram(left)and 2D electron density(right)for LiMn2O4
3 结 论
通过密度泛函理论第一性原理的计算方法研究LiMn2O4及其Ni掺杂材料的电子结构,所得主要结论如下:
1)LiMn2O4中Mn为嵌/脱锂过程中的氧化还原中心,而Ni掺入后Mn的价态保持不变,对于防止Mn的溶出有利,可保证材料结构的完整性;
2)Mn/Ni的3d轨道和O2p轨道之间具有很强的共价作用,而Li和O之间则为离子键作用;
3)过渡金属和氧之间的强相互作用使得晶格在锂离子嵌入/脱出过程中能够保持较好的结构完整性,使得材料的充放电过程为可逆的过程。
