液相色谱系统峰对煤基油品烃族组成的影响
2018-10-15李军芳
李 军 芳
(1.煤炭科学技术研究院有限公司 煤化工分院,北京 100013;2.煤炭资源高效开采与洁净利用国家重点实验室,北京 100013)
0 引 言
煤基油品中烃类化合物占95%~98%,烃类化合物的组成及结构决定了煤基油品的性质,对油品评价及后续提质加工工艺控制非常重要,详细的烃类组成分析也是油品加工工艺设计的基础数据[1-3]。高效液相色谱法(HPLC)具有样品用样量少、操作步骤简单、分析测试耗时短等优点,广泛应用于油品烃族含量的测定[4-7]。在建立煤基油品烃族组成测定方法的过程中,液相色谱不进样的情况下,进样阀或柱空切换后,在所收集的色谱图上有一些正峰或负峰,这些正峰或负峰被称为系统峰[8-9],系统峰的存在一定程度上对被测样品峰产生干扰。Levin 等[10-11]研究了液相色谱系统峰的起源、形成及重要性。叶利民等[12]对高效液相色谱柱切换技术中系统峰的影响进行考察,对流动相组成、预柱长短及填料种类等因素与系统峰的关系进行单因素变换分析,提出了避免系统峰对被测组分干扰的办法。龚时琼[13]分析了高效液相色谱分析中的异形峰,并提出相应的处理方法,但未讨论系统峰与异形峰的关联,关于异形峰对烃类组成测定的影响鲜见文献报道。为了规避液相色谱系统峰对煤基油品烃族组成的测定影响,笔者采用高效液相色谱正相色谱模式,固定相选用NH2柱,流动相采用正庚烷试剂,考察了一系列饱和烃标准物质的进样情况,采用混合饱和烃标准物质进行进样分析,利用不同标准物质保留时间微小的差异相互抵消可有效规避异形峰,且考虑到不同标准物质响应因子的差异,使测试结果更趋于真值。
1 试 验
1.1 试验仪器
高效液相色谱仪配备:在线脱气包,Waters1525高压输液泵,Waters 2707自动进样器,Waters 2414示差折光检测器,Waters NH2柱及保护柱和可温控的柱温箱,Breeze色谱数据处理工作站等。
1.2 试验试剂
正庚烷,分析纯,经过滤脱气后使用;正辛烷(C8)、正壬烷(C9)、正十六烷(C16)、甲基环己烷、邻二甲苯、1-甲基萘、芴、1-甲基菲、芘均为色谱纯。
1.3 色谱条件
流动相:正庚烷分析纯试剂;流速控制为0.6 mL/min;进样量为6 μL;固定相色谱柱:Waters Spherisorb®5.0 μm NH2(4.6 mm×250 mm);柱温设定为30 ℃;检测器为示差折光检测器(温度设定至35 ℃)。
1.4 色谱柱的选择
液相色谱分析油品烃类组成是利用色谱柱固定相与油品各组分间的吸附强弱进行分离。石油基油品的烃族组成测定现行标准[14-15]中大多利用正向色谱。色谱柱选用氰基柱或氨基柱。氨基柱是根据烃类物质的双键数和缩合芳环数进行分离,其取代基对容量因子的影响较小。苯、四氢萘与八氢萘将出现在同一洗脱位置,原因是氨基柱中具有相同双键数的分子作为同一个族组分被同时洗脱出,形成同一个谱峰。具有相同双键数的组分内,不同分子量的化合物对其保留时间影响较小[16]。因此,选用氨基柱,煤基油品组分能得到较好的分离效果。
1.5 保护柱的选择
由于杂原子的存在,煤基油品中会有极性化合物,而氨基柱对极性化合物的吸附不可逆,试验中需通过反冲洗才能淋洗除去极性物,但经常反冲色谱柱又会影响色谱柱柱效。为了维持分析色谱柱的柱效,延长分析色谱柱的使用寿命,通过在氨基柱前加装保护柱,减少对分析色谱柱的污染。
1.6 流动相的选择
液相色谱正相色谱系统中流动相一般采用非极性溶剂,如正己烷或正庚烷溶液。正己烷的沸点69 ℃,正庚烷的沸点98.5 ℃,且正庚烷的溶解能力更强,从实验室环境的安全性考虑,采用正庚烷作为流动相。色谱标样及样品都采用与流动相一致的正庚烷试剂溶解。
2 结果与讨论
2.1 系统峰的影响
2.1.1 拖尾峰
色谱图上饱和烃出峰位置(4.33~4.83 min)易出现异形峰,C8、C9、C12饱和烃标准物质色谱如图1所示。可知,采用正辛烷(C8)、正壬烷(C9)、正十二烷(C12)为饱和烃标准物质时,在饱和烃出峰位置出现拖尾小峰。进样C8和邻二甲苯时饱和烃出峰位置产生拖尾小峰(图1(a)),进样C9时产生拖尾小峰(图1(b)),进样C12、邻二甲苯和1-甲基萘时饱和烃出峰位置产生拖尾小峰(图1(c))。
2.1.2 峰型完好
当采用环己烷作为饱和烃标准物质,色谱图中在饱和烃出峰位置,峰型完好,进样环己烷时形成的色谱峰如图2所示。
2.1.3 前伸峰
当饱和烃标准物质选用正十六烷(C16)或甲基环己烷,色谱图中在饱和烃出峰位置则出现前伸的小峰。进样C16和邻二甲苯时饱和烃出峰位置产生的前伸峰(图3(a)),进样甲基环己烷时产生前伸峰(图3(b))。

图1 C8、C9、C12饱和烃标准物质色谱图Fig.1 Chromatogram of C8,C9,C12 as saturated hydrocarbon standard substance

图2 环己烷作为饱和烃标准物质色谱图Fig.2 Chromatogram of cyclohexane as saturated hydrocarbon standard substance
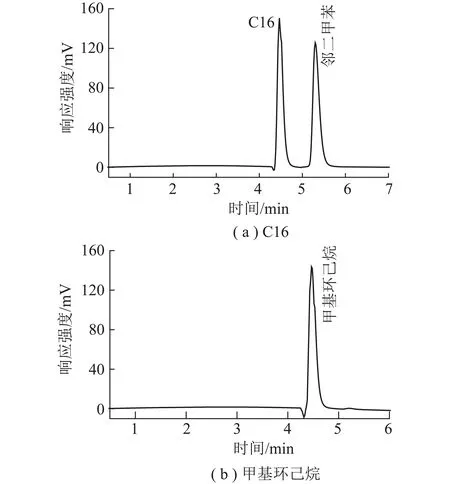
图3 C16、甲基环己烷作为饱和烃标准物质色谱图Fig.3 Chromatogram of C16 and methyl cyclohexane as saturated hydrocarbon standard substance
2.1.4 倒峰现象
采用单纯的流动相正庚烷作为样品进样,在色谱图中饱和烃出峰位置出现倒峰(图4),改变流速可改变倒峰的出峰时间,但不会使倒峰消失。
根据上述试验现象分析,倒峰为系统倒峰,系统峰产生原因可能是由于进样时系统扰动,导致柱平衡的弛豫过程[17]。系统峰与样品无关,独立于样品而存在,往往出现在色谱柱的死体积附近,通常形成正向或倒向的色谱峰。上述异形峰产生的原因是由于系统倒峰的存在,保留时间稍有差异的饱和烃试样正峰与系统倒峰相互抵消产生的结果。
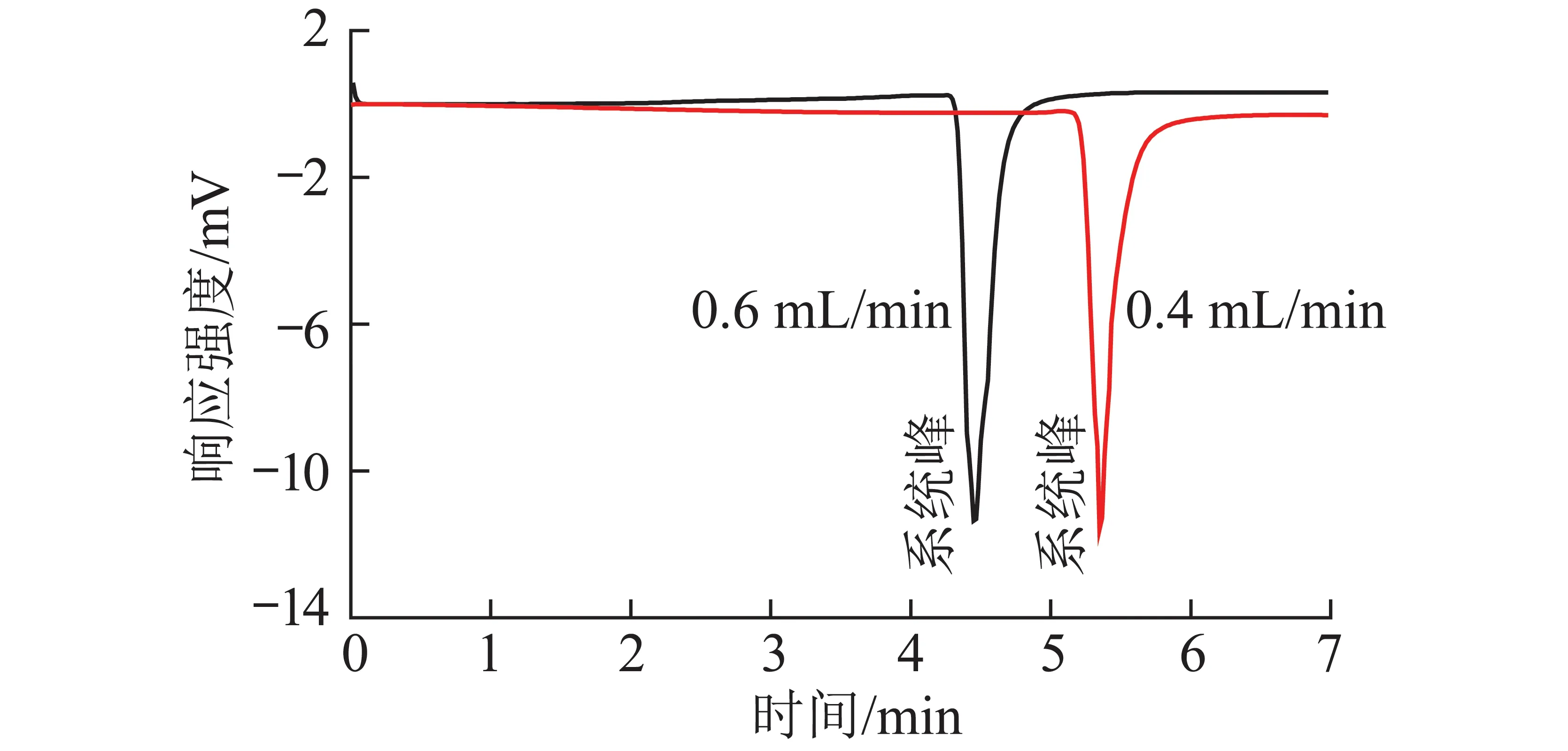
图4 流动相正庚烷作为样品直接进样色谱图Fig.4 Chromatogram of mobile phase n-heptane as sample direct injection
试验过程中通过改变流动相流速、流动相换用色谱纯正庚烷、改变柱温和进样方式等,系统倒峰的保留时间和峰型有所变化,但无法规避系统倒峰。此液相系统中,在不改变流动相组成及配比,即不改变流动相极性的前提下,无法将饱和烃出峰位置与系统倒峰出峰位置分离开,原因是固定相采用氨基柱,流动相需为非极性,而待分析的饱和烃物质也是非极性。该色谱系统中待分析的饱和烃组分在色谱柱中基本没有保留直接流出,造成系统倒峰和试样中饱和烃组分正峰无法分离。SH/T 0806—2008《中间馏分芳烃含量的测定 示差折光检测器高效液相色谱法》及SN/T 2380—2009《石油产品中芳烃含量的测定 高效液相色谱法》中并未考虑及说明系统倒峰对饱和烃出峰的影响,推测原因是由于所采用液相色谱柱柱效不高,微小的保留时间差异无法显现,样品出峰面积将系统倒峰面积完全掩盖。改变流动相极性能使饱和烃出峰位置与系统倒峰出峰位置分开,但系统倒峰后移又会影响后续芳烃的出峰面积计算,且不同饱和烃的标准物质响应值差异较大。考虑采用混合标准物质进行后续标定,既可将系统倒峰完全规避抵消(标准物质、样品积分面积都有内部抵扣),又使测试结果更趋于真值。
2.2 响应因子的考察
文献[18]表明族组成测定过程中,其余各族烃化合物的响应值差异很小,而饱和烃中各链烷烃和环烷烃的响应值差异很大。利用实验室现有的标准物质考察各族烃化合物的响应因子,结果见表1。
表1各族标准物质的保留时间和响应因子
Table1Retentiontimeandresponsefactorofstandardcompounds

族组成标样名称保留时间/min响应因子/10-5正辛烷4.636.79正壬烷4.676.03饱和烃正十二烷4.685.89正十六烷4.834.92环己烷4.759.39甲基环己烷4.786.86苯5.472.81一环芳烃邻二甲苯5.432.43茚满5.382.61二环芳烃1-甲基萘6.931.21芴系芴8.021.13三环芳烃1-甲基菲10.051.15四环芳烃芘11.831.06
由表1可知,芳烃中一环芳烃标准物质的响应因子接近,且二环芳烃、芴系、三环芳烃以及四环芳烃的标准物质响应因子很相似。然而饱和烃中链烷烃和环烷烃的响应因子有很大不同,随着碳数的增加,链烷烃标准物质的响应因子逐渐减小,随着侧链的增加,环烷烃标准物质的响应因子也逐渐递减。
2.3 标准物质的选定
对于煤基油品液相色谱烃族组成的测定,根据油品沸程的不同,不同馏分段采用不同标准物质进行外标曲线法测定。参照待分析样品质谱烃组成进行油品液相色谱混合标准物质的选取,最好选择实验室半制备柱制备的待测油分各烃族混合物作为液相色谱标样。为了操作方便,混合标准物质可按照等比例混配。不同馏分油的标准物质选择见表2。
表2不同馏分油的标准物质选择
Table2Selectionofdifferentdistillatesstandardsubstance

标样FBP<170 ℃FBP<300 ℃FBP<400 ℃饱和烃正辛烷正十二烷正十二烷、正十六烷甲基环己烷甲基环己烷甲基环己烷一环芳烃邻二甲苯邻二甲苯邻二甲苯二环芳烃—1-甲基萘1-甲基萘芴系—芴芴三环芳烃——1-甲基菲四环芳烃——芘
注:FBP为终馏点。
对煤基中间馏分油(相当于柴油馏分)进行分析测定,根据表2选择FBP<400 ℃的对应标准物质,采用正十二烷、甲基环己烷和正十六烷按照等比例混配,作为饱和烃的混合标准物质。色谱标准物质分离谱图和样品的分离色谱图如图5所示。由图5可知,采用饱和烃混合标样,饱和烃出峰位置,峰型完整,峰面积积分数据可靠。由于样品饱和烃本身是混合物,所以饱和烃出峰位置峰型完好。但样品中芳烃化合物成分相对复杂,各族之间相互干扰,致使二环芳烃和芴系化合物谱峰不能很好分开。

图5 标样和煤基油品的分离谱图Fig.5 Chromatogram of standard substance and coal-derived oil
2.4 方法的精密度和准确度试验
2.4.1 精密度试验
对同一个烃族组成的混合掺比样品测定5次,结果见表3,样品烃含量计算方法参照文献[18]。可知,由于仪器稳定性较好,该方法重复性较高,相对标准偏差均在3%以下,精密度较高。
2.4.2 准确度试验
用标准物质制备不同比例的已知各烃族组成的混合掺比样品(命名为掺混样品1号、2号、3号),在相同色谱条件下进行准确度空白加标回收率试验,结果见表4。由表4可知,空白加标回收率能控制在94%~105%,准确度满足色谱定量的要求。
表3精密度试验
Table3Precisionexperiments

族组成烃含量/%第1次第2次第3次第4次第5次平均值标准偏差相对标准偏差/%饱和烃30.5231.1231.0830.5730.9930.860.293 5 0.95一环芳烃34.8434.6634.2634.3133.334.270.595 7 1.74二环芳烃22.1822.222.2123.0123.3622.590.555 4 2.46芴系5.305.015.345.095.165.180.139 1 2.69三环芳烃5.085.004.974.895.014.990.068 9 1.38四环芳烃2.082.012.142.132.182.110.056 1 2.67
表4准确度试验
Table4Accuracyexperiments

样品族组成理论值/%实测值/%回收率/%饱和烃35.0534.8599.43一环芳烃34.0635.06102.94掺混样品1号二环芳烃20.4120.0998.43芴系2.051.9595.12三环芳烃4.123.8994.42四环芳烃4.314.1696.52饱和烃60.0259.7699.57一环芳烃20.0020.73103.65掺混样品2号二环芳烃12.5011.9695.68芴系4.204.1699.05三环芳烃1.871.94103.74四环芳烃1.411.45102.84饱和烃75.0074.6099.47一环芳烃15.0515.15100.86掺混样品3号二环芳烃5.205.27101.35芴系1.751.86104.57三环芳烃1.461.53104.79四环芳烃1.541.59103.25
2.4.3 液相色谱与质谱法对比
为进一步验证试验液相色谱方法的可靠性,对同一样品采用高效液相色谱法与质谱法测试结果进行对比,几种典型煤基中间馏分样品(命名为1~4号)的族组成试验结果见表5。由表5可知,2种不同分析手段的饱和烃相对偏差最大为2.29%,总芳烃相对偏差最大为4.12%,2种方法测试结果的相对偏差不超5%,证明该方法的准确性较高。
煤基石脑油的杂原子含量低,尤其加氢后煤基石脑油杂原子含量极低,文献[18]测定煤基石脑油不考虑极性化合物的存在,对测试结果进行归一化处理可行。值得注意的是,煤基油品中间馏分(柴油馏分)硫、氮、氧等杂原子含量高时,必须考虑极性化合物的存在,此时不能简单进行归一化处理,而要利用外标曲线测试结果进行差减计算得到极性化合物含量。对于深度加氢后的煤基柴油馏分,由于杂原子基本脱除,可不用考虑极性化合物存在,直接进行归一化处理。
表52种分析方法试验结果
Table5Experimentalresultsoftwoanalysismethods%
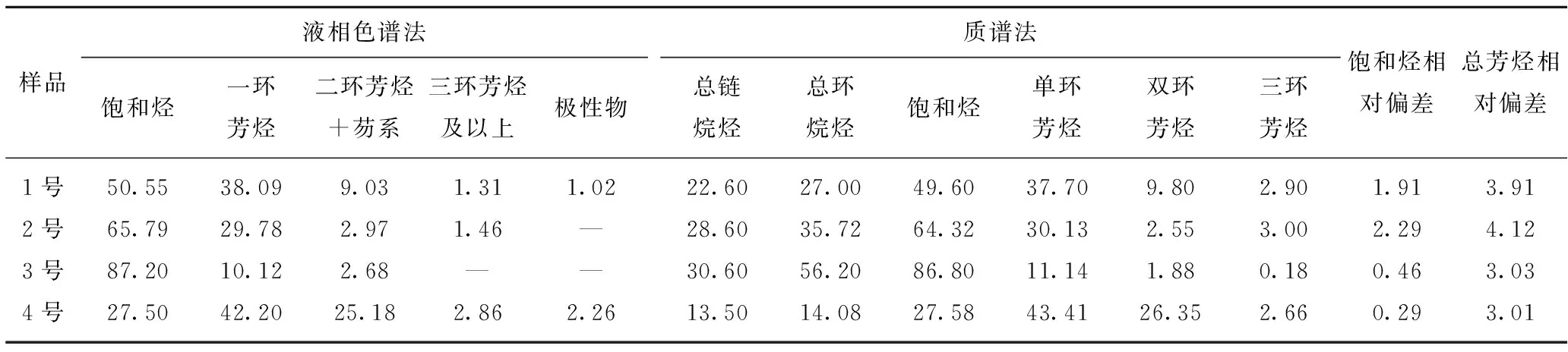
样品液相色谱法饱和烃一环芳烃二环芳烃+芴系三环芳烃及以上极性物质谱法总链烷烃总环烷烃饱和烃单环芳烃双环芳烃三环芳烃饱和烃相对偏差总芳烃相对偏差1号50.5538.099.031.311.0222.6027.0049.6037.709.802.901.913.912号65.7929.782.971.46—28.6035.7264.3230.132.553.002.294.123号87.2010.122.68——30.6056.2086.8011.141.880.180.463.034号27.5042.2025.182.862.2613.5014.0827.5843.4126.352.660.293.01
3 结论与建议
1)对于氨基柱为固定相,正庚烷为流动相的正相色谱模式,系统倒峰出峰位置无法与样品中饱和烃出峰位置分开;但当饱和烃标准物质采用混合标准物质时,既规避了系统峰的干扰,又使测试结果更趋于真值。
2)高效液相色谱法是快速准确分析油品族组成的有效手段,建立的液相色谱煤基油品测定方法,准确度回收率在94%~105%;液相色谱与质谱法结果对比,饱和烃与总芳烃相对偏差均不超5%。
3)根据油品馏程不同,采用不同标准物质建立多条测定曲线,使得测量结果更为可靠。建议最好选择实验室半制备柱制备的待测油分各烃族混合物作为液相色谱标样。
4)当待测组分硫、氮、氧等杂原子含量高时,必须考虑极性化合物的存在,此时对测试的结果不能简单进行归一化处理,而要利用外标曲线测试结果进行差减计算得到极性化合物的含量。
