转录组学在茶树研究中的应用进展
2018-04-18江新凤刘本英李友勇李洪波
江新凤 刘本英 李友勇 李洪波
(1.江西省蚕桑茶叶研究所 330202;2.云南省茶学重点实验室 666201)
中国是茶的故乡,茶产业的发展源远流长,茶树种质资源丰富,截至2017年,通过国家级审(认、鉴)定的茶树品种有134个,数量位于世界前列[1~3]。长期以来,对于茶树种质资源以及品种特性研究均通过形态学特征和农艺特征观察来实现,但由于茶树是一种长期异化授粉、自交不亲和性的作物,表现为高度杂合的生殖特点,加上品种选育均采用传统的系统和杂交选育,且父母本亲缘关系较近,其后代发生的遗传变异小,故通过表型来进行品种鉴定存在一定的难度和不确定性。
转录组学为茶树基因研究开辟了新途径,对茶树的进化、基因突变以及翻译调控等多种生物学过程提供有效工具,也可作为标记进行辅助育种,降低标记选择成本,缩短了育种进程[4~6]。本文对近年来茶树转录组学研究进展进行总结,为转录组测序技术在茶树上的进一步应用提供参考。
1 转录组学概况
转录组学(Transcriptomics)是研究细胞在某一功能状态下所含mRNA的类型与拷贝数;比较不同功能状态下mRNA表达的变化,搜寻与功能状态变化紧密相关的重要基因群,对每种转录本在发育过程中和不同条件下表达水平的变化进行量化[7~8]。转录组的概念是由Velculescu等人[9~10]于1997年最早提出的,20 世纪90 年代中期,转录组学开始在生物学领域作为一门新技术,成为研究热点并且得到广泛的应用[11~12],比如EST(Expressed Sequence tags,表达序列标签 )是从已建好的cDNA库中随机抽取克隆,从5’末端或3’末端对插入的cDNA片段进行一轮单向自动测序,所获得的约60~500bp的一段cDNA序列。20世纪90年代初Craig Venter提出了EST的概念,并测定了609条人脑组织的EST,宣布了cDNA大规模测序时代的开始[13]。1993年前EST数据收录于GenBank、EBI和DDBJ。同年NCBI(National Center of Biotechnology Information)建立了一个专门的EST数据库dbEST来保存和收集所有的EST数据。1995年中期GenBank中EST的数目超过了非EST的数目。现在GenBank中EST的数目已经超过了三千五百万,约占GenBank中序列数的60%。转录组学方法尤其是cDNA微阵列的应用促进基因表达数据爆炸性增长,但如何对这些数据进行分析,从中提取有意义的生物学信息,是未来一段时间转录组学的研究热点和需要攻克的技术瓶颈。
2 转录组学的研究方法
随着科学研究的不断深入,杂交技术、PCR 技术、标签序列分析等新方法、新技术已成功地应用于转录组学研究。转录组学研究方法到目前为止,经历三代发展。早期,由于测序价格昂贵、基因序列数目有限,转录组学研究者只能进行极少数特定基因的结构功能分析和表达研究。最早广泛应用测序技术为70年代的 Sanger 法,这也是完成人类基因组计划的基础。最近十几年, 分子生物学技术的快速发展使高通量分析成为可能,高通量研究方法主要可以分为两类:一类是基于杂交的方法,主要是指微阵列技术(Microarray);一类是基于测序的方法,这类方法包括表达序列标签技术(Expression Sequence Tags Technology,EST)、基因表达系列分析技术(Serial analysis of gene expression,SAGE)、大规模平行测序技术(Massively parallel signature sequencing,MPSS)、RNA 测序技术(RNA sequencing,RNA-seq)。自2005年以来,以Roche 公司的454技术、Illumina 公司的 Solexa 技术以及 ABI 公司的 Solid 技术为标志的高通量测序技术相继诞生[14~15]。不同转录组研究方法比较见表1[16~17]。转录组学研究有助于了解特定生命过程中相关基因的整体表达情况,进而从转录水平初步揭示该生命过程的代谢网络及其调控机理。随着后基因组时代的到来,转录组测序成为率先发展且应用相对广泛的技术 。
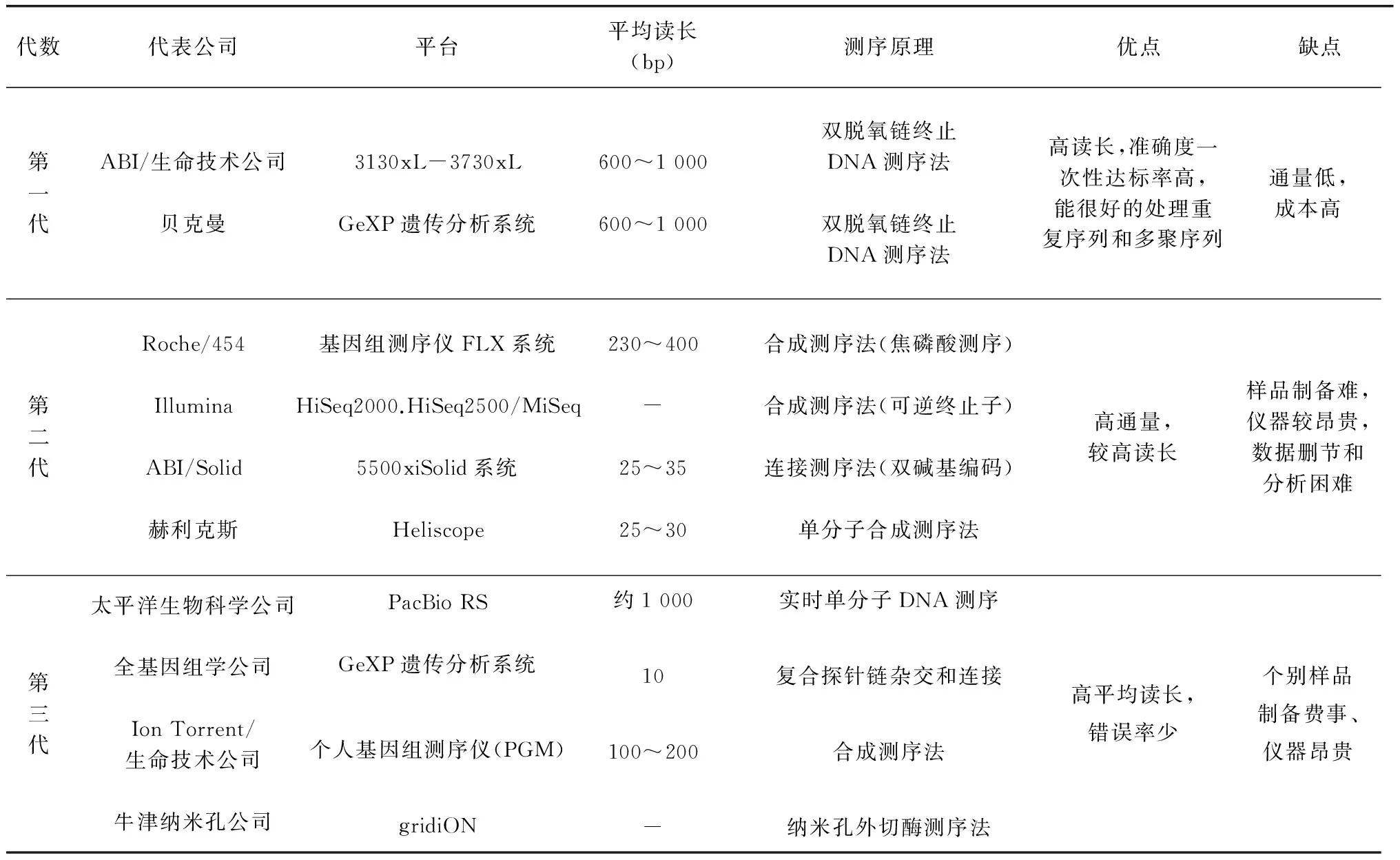
表1 各种转录组(测序技术)研究方法比较
3 转录组学在茶树研究中的应用
高通量测序技术的发展极大推动了转录组学的研究,使研究者能发现更多新转录本,挖掘更多分子标记,更清晰地绘制转录图谱以及更准确地确定代谢途径通路[6]。转录组技术在茶树中的应用较玉米、大豆、水稻等大田作物较少[18]。但是随着第三代测序技术的不断完善和发展,将来测序成本会大大降低,测序的通量和准确性也会不断提高,其在茶树的功能基因挖掘、次生代谢调控、抗性及茶树遗传育种和进化分析等研究中将进一步拓展。
3.1 功能基因挖掘
目前,由于基因组测序的功能注释还不够完备,因此基因优化可以通过转录组测序技术进行,通过深入比较分析已知基因组注释模型与转录组测序结果,从而挖掘该物种的新基因及完善其基因组的注释[19]。张成才[20]以中茶108和福鼎大白茶为材料,进行了自交(SP)和杂交授粉(CP),选取了自交和杂交授粉后24h、48h和72h的花柱,进行转录组学分析,结果发现,自交和杂交授粉花柱在基因表达模式上有显著差异,筛选到了大量可能与茶树育性相关的重要基因;韦康[21~22]使用Illumina测序法对茶树扦插枝条使用吲哚丁酸(IBA)进行转录组分析,发现许多基因参与了植物激素信号转导、次生代谢、细胞壁组织和谷胱甘肽代谢;谭立强[23]利用Illumina测序平台进行高通量测序,分析了茶树的转录组,构建SSR覆盖1 156.9cm的237个SSR标记分布的15个连锁群遗传连锁图谱;马春雷[24]利用第一代cDNA微阵列方法对“安吉白茶”泛白不同阶段基因表达进行了分析,筛选部分茶树功能基因;韦朝领[25]发现了茶树特有化合物主要代谢途径的大量候选基因。
3.2 次生代谢调控
近年,转录组技术在次生代谢调控方面运用颇多,对茶树特征性次生代谢相关基因进行挖掘,以及对基因的结构、功能、表达及调控方面研究也有相关报道[26]。李春芳[27]通过对茶树的13个不同组织部位进行转录组测序,得到了34.7万条特异的茶树基因序列,找到了1 719个基因参与次生代谢产物的合成,其中206个基因参与类黄酮、咖啡因和茶氨酸的合成,找到了339个可能调控类黄酮、咖啡因和茶氨酸合成途径基因的转录因子。邰玉玲[28]对茶树和油茶转录组数据进行比较分析,研究了茶树特征性代谢成分形成的分子机制,结果表明,在茶树特征性成分积累的过程中,与儿茶素、茶氨酸和咖啡碱相关的代谢途径的基因表达量茶树比油茶高;王璐[29]通过比较龙井43和中黄2号,分析了两品种的生化成分、叶绿体结构、基因表达与代谢途径,发现中黄2号与龙井43中存在4 902个差异基因,其中259个与氨基酸代谢、光合作用和色素代谢有关;吴华玲[30]用20个特色茶树品系的嫩梢、幼叶、成熟叶为材料,构建cDNA文库并利用Roche/454转录组测序,共读取437 908个基因序列,通过重组装得到25 637条,并用这些序列与公众数据库进行比对。结果发现这些基因的大多数映射到碳水化合物代谢、能量代谢和次生代谢产物生物合成途径。
3.3 抗性研究
刘声传[31]以“宁州2 号”为RNA-Seq材料进行了转录组测序,揭示了干旱胁迫与复水下茶树激素代谢和信号转导以及可溶性糖和脯氨酸代谢的机理;王璐[32]、王新超[33]、张悦[34]使用Illumina及RNA-Seq技术对茶树冷胁迫下进行全转录组测序,通过比较转录组或基因表达谱的研究以揭示生物学现象或疾病发生的分子机制。王丹[35~36]比较不同茶树品种转录组差异,借助生物信息学方法分析海量数据后,对茶树抵御茶尺蠖抗性相关基因进行了功能注释、代谢通路分析,同时关键基因进行了筛选。
4 展望
目前,转录组测序技术已在医学和农学等基础以及应用基础研究中广泛应用。由于测序技术和生物信息学的不断发展,分析其测序结果会越来越真实可靠,基于转录组数据的分析研究,将有助于得到新的功能基因和代谢通路,为茶树种质资源鉴定、保存与优良种质选育提供分子基础;通过对次生代谢途径关键酶基因的研究,为茶树活性成分的生物合成与调控提供新的思路和方法,或通过基因转录水平的调节,提高茶树功能成分的产量与活性,为茶树的良种选育、规范化种植和质量控制提供技术支撑。当然,作为一种快速、高通量、全面解读茶树基因组信息的全新技术手段,茶树转录组研究在基础理论和生产实践中均具有重要的意义的同时也存在一些弊端,如样本需求量大、容易受环境因素的影响,因此转录组学的应用结合众多新兴组学,例如蛋白质组学和代谢组学的研究,实现高通量与高效率的结合,揭示传统茶树生物学内涵,为茶产业的发展提供助力。
[1]江新凤,李文金,杨普香.江西省茶树种质资源研究进展[J].蚕桑茶叶通讯,2016(5):29~31.
[2]Kamunya S M, Wachira F N, Pathak R S, et al. Genomic mapping and testing for quantitative trait loci in tea (Camelliasinensis(L.) O. Kuntze) [J]. Tree Genetics & Genomes, 2010, 6(6): 915~929.
[3]Zhao L P, Liu Z, Chen L, et al. Generation and characterization of 24 novel EST derived microsatellites from tea plant (Camelliasinensis) and cross-species amplification in its closely related species and varieties [J]. Conservation Genetics, 2008, 9(5):1327~1331.
[4]Chen L, Zhou Z X, Yang Y J, et al. Genetic improvement and breeding of tea plant (Camelliasinensis) in China: from individual selection to hybridization and molecular breeding [J]. Euphytica, 2007, 154:239~248.
[5]史硕博,陈涛,赵学明.转录组平台技术及其在代谢工程中的应用[J].生物工程学报,2010,26(9):1 187~1 198.
[6]王继玥,余庭跃,张彩波.玉米转录组学研究进展[J].华北农学报,2014,29(增刊):10~15.
[7] Wang Z, Gerstein M, Snyder M, et al. RNA-Seq: a revolutionary tool for transcriptomics[J]. Nat Rev Genet, 2009,10(1):57~63.
[8] Costa V, Angelini C, De F I, et al. Uncovering the complexity of transcriptomes with RNA-Seq [J]. Journal of Biomedicine & Biotechnology, 2010, 5 757 (2 010):e853916.
[9] Velculescu V E, Zhang L, Zhou W, et al. Characterization of the yeast transcriptome[J]. Cell, 1997, 88(2):243~251.
[10] 徐云碧.分子植物育种[M].北京:科学出版社,2014:79~80.
[11] Lockhart D J,Winzeler E A. Genomics,gene expression and DNA arrays[J].Nature,2000, 6 788 (405): 827~836.
[12] Birzele F,Schaub J,Rust W,et al.Into the unknown: expression profiling without genome sequence information in CHO by next generation sequencing[J].Nucleic Acids Research,2010,38 ( 12 ) :3 999~4 010.
[13] Adams M D, Kerlavage A R, Fleischmann R D, et al. Initial assessment of human gene diversity and expression patterns based upon 83 million nucleotides of cDNA sequence[J]. Nature, 1995, 6 547 (377,Suppl):3.
[14] Maher C A, Kumarsinha C, Cao X, et al. Transcriptome sequencing to detect gene fusions in cancer[J]. Nature, 2009, 7 234 ( 458):97~101.
[15] Zhou X, Ren L, Li Y, et al. The next-generation sequencing technology: a technology review and future perspective.[J]. Science China Life Sciences, 2010, 53(1):44.
[16]杨晓玲,施苏华,唐恬.新一代测序技术的发展及应用前景[J].生物技术通报,2010(10):76~82.
[17]田李,张颖,赵云峰.新一代测序技术的发展和应用[J]. 生物技术通报,2015,31(11):1~8.
[18]刘冠,赵婷婷,杨欢欢,等.番茄转录组学研究进展[J].基因组学与应用生物学,2016,35(10):2 802~2 807.
[19] 窦孝锐.马铃薯转录组测序研究进展[J].现代农业科技,2015(13):81~83.
[20] Zhang C C, Wei K,Ni D J, et al. Transcriptome analysis reveals self-incompatibility in the tea plant (Camelliasinensis) might be under gametophytic control [J]. BMC Genomics,2016,17(1):359.
[21] Wei K, Wang L Y, Wu L Y, et al. Transcriptome analysis of indole-3-butyric acid-induced adventitious root formation in nodal cuttings ofCamelliasinensis(L.)[J]. Plos One, 2014, 9(9):e107201.
[22] Wei K, Wang L, Zhang C, et al. Transcriptome analysis eveals key flavonoid 30-hydroxylase and lavonoid 30,50-hydroxylase genes in affecting the ratio of dihydroxylated to trihydroxylated catechins inCamelliasinensis. [J].Plos One 2015,10(9): e0137925.
[23]Tan L Q, Wang L Y, Wei K, et al. Floral transcriptome sequencing for SSR marker development and linkage map construction in the tea plant (Camelliasinensis)[J]. Plos One, 2013, 8(11):e81611.
[24]Ma C L, Chen L, Wang X C,et al. Differential expression analysis of different albescent stages of Anji Baicha (Camelliasinensis(L.) O. Kuntze) using cDNA microarray[J]. Scientia Horticulturae ,2012,148:246~254.
[25] Shi C Y, Yang H, Wei C L, et al. Deep sequencing of theCamelliasinensistranscriptome revealed candidate genes for major metabolic pathways of tea-specific compounds[J]. BMC Genomics, 2011, 12(1):131.
[26]宛晓春,夏涛.茶树次生代谢[M].北京:科技出版社,2015:180.
[27] Li C F, Zhu Y, Yu Y, et al. Global transcriptome and gene regulation network for secondary metabolite biosynthesis of tea plant (Camelliasinensis)[J]. BMC Genomics, 2015, 16(1):560.
[28] Tai Y L,wei C L,Wan X C,et al.Transcriptomic and phytochemical analysis of the biosynthesis of characteristic constituents in tea (Camelliasinensis) compared with oil tea (Camelliaoleifera)[J]. BMC Plant Biology,2015, 15(1): 190.
[29] Wang L, Yue C, Cao H, et al. Biochemical and transcriptome analyses of a novel chlorophyll-deficient chlorina teaplantcultivar[J]. BMC Plant Biology, 2014, 14(1):352.
[30] Wu H L, Chen D, Li J, et al. De Novo characterization of leaf transcriptome using 454 sequencing and development of EST-SSR markers in tea (Camelliasinensis)[J]. Plant Molecular Biology Reporter, 2013, 31(3):524~538.
[31] Liu S C, Jin J Q, Ma J Q, et al. Transcriptomic analysis of tea plant responding to drought stress and recovery[J]. Plos One, 2016, 11(1):e0147306.
[32] Wang L, Wang X, Yue C, et al. Development of a 44 K custom oligo microarray using 454 pyrosequencing data for large-scale gene expression analysis ofCamelliasinensis[J]. Scientia Horticulturae, 2014, 174(1):133~141.
[33] Wang X C, Zhao Q Y, Ma C L, et al. Global transcriptome profiles ofCamelliasinensis, during cold acclimation[J]. BMC Genomics, 2013, 14(1):415.
[34] Zhang Y, Zhu X, Chen X, et al. Identification and characterization of cold-responsive microRNAs in tea plant (Camelliasinensis) and their targets using high-throughput sequencing and degradome analysis[J]. BMC Plant Biology, 2014, 14(1):271.
[35] Wang D, Li C F, Ma C L, et al. Novel insights into the molecular mechanisms underlying the resistance ofCamelliasinensisto Ectropis oblique, provided by strategic transcriptomic comparisons[J]. Scientia Horticulturae, 2015, 192(2):429~440.
[36]王丹,陈亮.茶树对茶尺蠖抗性机制研究[J].茶叶科学,2014,34(6):541~547.
