微生态制剂调节便秘、腹泻人群肠道菌群结构与产短链脂肪酸关键菌属的相关性
2018-03-20臧凯丽陈庆森赵林森崔文静马新颖闫亚丽
臧凯丽,江 岩,孙 勇*,陈庆森,赵林森,赵 培,*,崔文静,马新颖,闫亚丽
(1.天津商业大学生物技术与食品科学学院,天津市食品生物技术重点实验室,天津 300134;2.中国肉类食品综合研究中心,北京食品科学研究院,北京 100068;3.河北一然生物科技有限公司,河北 石家庄 050800)
肠道菌群的基因组信息被称为人类的“第二基因组”,编码了330万 个非冗余的基因,大约是人类基因编码能力的150 倍[1],拥有丰富的遗传信息,参与人体营养物质的消化吸收,调控多种代谢途径和机体的免疫系统,故与机体健康息息相关。近些年来研究显示,宿主和微生物基因组共同调节并通过产生短链脂肪酸(shortchain fatty acids,SCFAs)、胆汁酸、胆碱以及吲哚等代谢物质维持机体健康[2],其中乙酸、丙酸、丁酸相对含量较高,约占SCFAs总量的90%以上[3],对人体结直肠的健康有重要作用[4-6]:丁酸或产丁酸盐的肠道细菌具有恢复宿主免疫功能,保护屏障完整性和调节能量代谢等作用,丁酸盐会影响中性粒细胞的功能和转移,抑制血管细胞的炎性细胞因子,增加结肠上皮细胞紧密连接蛋白表达,并通过人体免疫细胞减少细胞因子和趋化因子表达,其他SCFAs如丙酸和乙酸通过血液作用于各种不同的器官,被用作底物的氧化、脂质合成和肝细胞代谢的能量;此外,SCFAs还具有调节组蛋白去乙酰酶抑制剂的功能,刺激交感神经系统从而影响啮齿动物的社会行为,SCFAs也能刺激肠道蠕动和运输,并增加生理诱导浓度,在体外模拟结肠黏膜系统中5-羟色胺的释放增加8~10 倍。目前,慢性便秘、腹泻等胃肠道功能性疾病影响着全球2%~34%的社会人群,我国便秘的患病率已高达15.2%[7-8]。研究表明,饮食结构和营养的摄入是塑造肠道菌群结构的最主要因素,一方面其会改变肠道菌群的结构;另一方面食品中难以被人体吸收的成分则通过肠道菌群的作用发挥营养作用[9]。到目前为止,越来越多的研究表明,微生态制剂对人体益生功效已经受到广泛关注,这不仅体现在其可与病原体竞争结合位点和底物,维系宿主肠道微生物区系的平衡来促进机体肠道健康;还能够影响肠道内微生物和宿主代谢产物的分泌,例如代谢酶类、细胞因子和SCFAs,从而降低肠道内环境、提供能量、减少细菌易位和减轻内毒素血症等[10-12]。杨远志等[13]研究表明,饲喂抗性淀粉和乳酸菌的混合制品在降低结肠肿瘤方面的效果要优于单独使用益生素的效果。郭壮[14]应用实时荧光定量聚合酶链式反应(quantity polymerase chain reaction,qPCR)技术和测序技术综合分析评价乳酸菌在受试人群肠道的存活定植以及该菌株对受试人群肠道菌群多样性的影响,发现服用微生态制剂能够有效改善人体肠道菌群的组成,促进有益菌的生长,抑制致病菌及条件致病菌的繁殖;同时还有研究发现当受试人群停止服用微生态制剂后,其肠道菌群又恢复到服用前的结构[15]。本实验室自2013年以来,已利用Ion torrent PGM测序平台分别开展了益生菌和益生元对小鼠菌群结构以及代谢产物的相关课题,分析了瑞士乳杆菌Lactobacillus helveticus干预对小鼠肠道菌群结构的影响[16],探讨了魔芋葡甘露聚糖对溃疡性结肠炎小鼠肠道菌群的调节作用[17]。尽管微生态制剂表现出更好地维护肠道内环境稳态和增强机体健康的功效,但促进肠道关键菌群或菌种的研究工作还需进一步深入探究,以便更好地服务于人类健康。
基于微生态制剂具有通过调节肠道菌群结构来维持机体与肠道菌群动态平衡的作用,加之一些学者开展了水苏糖(stachyose tetrahydrate,Sta)、植物乳杆菌、嗜酸乳杆菌、干酪乳杆菌、两歧双歧杆菌功效的研究[18-21];因此,本研究以便秘和腹泻人群为对象,在不同微生态制剂的干预下,通过对受试者粪便DNA提取,利用Ion torrent PGM测序平台探究受试者肠道菌群结构信息的变化,同时利用气相色谱检测粪便中的SCFAs表达水平,解析微生态制剂干预前后肠道菌群多样性的变化及其与SCFAs代谢相关的关键菌属的相关性,比较Sta、益生菌纯粉(probiotics power,PP)以及益生菌剂(probotic preparations,PPrs)在调节肠道菌群结构和代谢SCFAs产物方面的能力,研究结果将为鉴定并阐述微生态制剂解决便秘和腹泻相关的肠道关键菌属的关系提供科学数据,也为通过微生态制剂以肠道菌群为靶点来调整、治疗便秘和腹泻提供新的途径。
1 材料与方法
1.1 材料、菌株与试剂
Sta(含85%水苏糖、麦芽糊精约10%~15%)、PP(含植物乳杆菌LP45(Lactobacillus plantarum)、嗜酸乳杆菌La28(Lactobacillus acidophilus)、麦芽糊精,LP45与La28活菌数比例为3∶1,活菌数3×1010CFU/g)、PPrs(含抗性糊精、菊粉、水苏糖、麦芽糊精、植物乳杆菌LP45(Lactobacillus plantarum)、嗜酸乳杆菌La28(Lactobacillus acidophilus)、干酪乳杆菌YMC1069(Lactobacillus casei)、两歧双歧杆菌TMC3115(Bifidobacterium bifidum),活菌数2×1010CFU/g)河北一然生物科技有限公司。
Ion Xpress™ Barcode Adapters Kit、Ion PGM™Template OT2 200 Kit、Ion PGM™ Sequencing 200 Kit v2、Ion Plus Fragment Library Kit、Ion Library TaqMan™Quantitation Kit、Pfu DNA聚合酶 美国Life-Thermo Scientific公司;Qubit®dsDNA HS Assay Kit 美国Invitrogen公司;High Sensitivity DNA Kit 美国Agilent公司;QIAamp Fast DNA Stool Mini Kit 德国QIAGEN公司;MiniBEST Agarose Gel DNA Extraction Kit 宝生物工程(大连)有限公司;SCFAs(乙酸、丙酸、异丁酸、丁酸(均为色谱纯)) 天津光复研究所;其他试剂均为化学纯。
1.2 仪器与设备
3K18高速冷冻离心机 美国Sigma公司;DYY-2C电泳仪 北京六一仪器厂;508-U001 Ion torrent测序平台 美国Invitrogen公司;1005527 Ion One TouchTM2INS模板制备系统、8441-21 Ion One Touch™ ES模板富集系统、900 SERIES超低温冰箱、磁力架、2.0 Qubit荧光定量仪 美国Life-Thermo Scientific公司;2 1 0 0 生化分析仪、7 8 9 0 A气相色谱仪 美国Agilent公司;N8050200 GeneAmp PCR系统7900 美国ABI公司;II-3 Biowave DNA紫外-可见分光光度计英国柏诺公司。
1.3 方法
1.3.1 受试人群的分组及样本采集
研究中所有样本均采集自河北省石家庄市正定县,受试人群年龄为18~35 岁;便秘、腹泻受试人群的临床症状确定:通过询问受试者和其自身表述,按照临床医学给定的便秘、腹泻等症状确定。要求受试人群在开始实验的两个星期内未发生任何疾病,实验期间无使用抗生素等药物或接受治疗的情况;3 个月内未参与其他实验研究。
在2014年8~11月期间,共38 名受试人员,其中男性13 名,女性25 名。受试人群保持饮食及睡眠正常且无限制,每日早晚各服用微生态制剂一次,2 g/次(活菌数约3×1010CFU/g);干预期间记录饮酒、服药等特殊情况。在服用微生态菌剂之前统一采集粪便一次,干预中受试人群粪便样本每周采集2次,共采集4 周,在停止服用制剂后,采集志愿人员粪便样本2 次。除去3 个异常人员外,对其余受试人员进行样本采集,并将样本统一放置于液氮罐中保存,共采集粪便样本280 个。用QIAamp DNA Stool Mini Kit提取35 个人的约220 个样粪便细菌基因组DNA。用1%琼脂糖胶检测后,又根据细菌基因组完整性以及浓度等判断,约有180 个样本符合要求,保存备用。最后选择不同生理状态:健康(1男2女)、便秘(3女)、腹泻(2男4女)以及服用不同微生态制剂:对照组(Ctro,1男2女共3 人均为健康者)、水苏糖组(Sta,1男2女共3 人均为腹泻者)、益生菌纯粉组(PP,2女为便秘者,1女为腹泻者,共3 人)、益生菌剂组(PPrs,1男1女为腹泻者,1女为便秘者,共3 人)的12 个人共104 个样本进行高通量测序。
1.3.2 粪便中微生物总DNA的提取
迅速称量从-20 ℃冰箱取出的粪便样本0.2 g,利用QIAamp Fast DNA Stool Mini Kit提取粪便中微生物的总DNA,提取方法在试剂说明书的基础上做了改良。用Biowave紫外-可见分光光度计进行核酸质量检测,在260 nm和280 nm波长处分别测定吸光度,并用1%的琼脂糖凝胶电泳检测DNA的质量。
1.3.3 肠道菌群16S rRNA V3区的PCR扩增
利用细菌通用引物扩增16S rRNA V3区片段,上游引物F:5’-ACT CCT ACG GGA GGC AGC AG-3’和下游引物R:5’-TTA CCG CGG CTG CTG GCA C-3’。PCR反应体系(50 μL):5 μL 10×buffer(含20 mmol/L MgSO4)、1 μL 10 mmol/L的dNTP(dNTP终浓度为0.2 mmol/L),上、下游引物(10 μmol/L)各0.5 μL,10~25 ng肠道细菌基因组DNA,最后加入0.2 μL Pfu DNA聚合酶(2.5 U/μL),加无菌水至50 μL。为避免非特异性扩增采用降落PCR的方法进行扩增,反应程序为:95 ℃ 3 min,95 ℃ 1 min,70 ℃ 30 s,开始每个循环降0.5 ℃,72 ℃ 1 min,共15 个循环;95 ℃ 1 min,62 ℃ 30 s,72 ℃ 1 min,共10 个循环,72 ℃ 1 min,最后72 ℃ 10 min。PCR产物用3%的琼脂糖凝胶电泳检测并用MiniBEST Agarose Gel DNA Extraction Kit回收纯化,纯化后的扩增产物用Qubit®dsDNA HS Assay Kit进行PCR产物的定量分析。
1.3.4 Ion torrent PGM测序
首先,用Ion Plus Fragment Library Kit和Ion Xpress™Barcode Adapters Kit进行测序文库的构建。然后,进行测序模板的制备,Ion One Touch™ ES模板富集系统自动对携带有测序模板的Ion Sphere™ Particles进行富集。最后,在Ion torrent PGM测序平台上进行上机测序。
1.3.5 测序数据生物信息学和多变量统计学分析
首先,用FastQC软件将原始数据进行质控,并利用NGStool kits过滤掉低质量序列。将质控得到的高质量序列根据样本Barcode和引物进行数据的正、反方向分选,进入微生物生态学定量研究(quantitative insights into microbial ecology,QIIME)[22]后再次质控并去除嵌合体,利用usearch方法在97%的相似水平下划分分类操作单元(operational taxonomic units,OTUs);选取每个OTU中丰度最高的序列为代表序列,再参照样本的信息,生成以序列数代表的每个样本中每个OTU丰度的OTU Table。然后,依据OTU Table数据进行每个文库多样性分析,并利用R(版本3.1.3)软件绘制观测物种指数、Shannon多样性指数、Ace指数以及反映样本文库库容的Good’s coverage指数等。同样,以OTU Table为基础进行物种分类地位的确定,生成系统发育树,样本中各水平相对丰度划分并生成堆积图。最后,为了减少因某些样本中含有丰度极高的OTU而造成的样本间距离分布异常和测序时带来的偏差,研究中对所有样本中的OTUs丰度进行转化,转换后的数据即样本中各进化水平相对丰度百分比用R(版本3.1.3)绘制后续的丰度分布热图、堆积图并进行加权和非加权UniFrac距离矩阵的PCoA分析。
1.3.6 气相色谱分析粪便SCFAs
取粪样均质液2 mL,在12 000 r/min离心2 min后,取上清液1 mL,加入体积分数50%硫酸溶液200 μL振荡混匀15 s;再加入乙醚1 mL,再次涡旋振荡15 s,静置2 min进行萃取;室温下12 000×g离心5 min,转移上层有机相过滤膜,至一次性样品瓶中进行气相色谱分析。研究利用6890N气相色谱仪进行检测,配备极性的HP-FFAP色谱柱(30 m×0.25 mm,0.25 μm)以及氢火焰离子化检测器(f l ame ionization ditector,FID)检测器。检测的SCFAs标准品包括乙酸、丙酸、丁酸、异丁酸、戊酸和异戊酸共6 种。升温程序:初始柱温为140 ℃,持续10 min,以5 ℃/min的速率升温至165 ℃,再以25 ℃/min升至270 ℃,持续2 min。检测器温度为280 ℃,进样口温度为250 ℃。SCFAs保留出峰先后顺序为:乙酸、丙酸、异丁酸、丁酸、异戊酸、戊酸。
2 结果与分析
2.1 肠道菌群16S rRNA V3区测序文库的检测
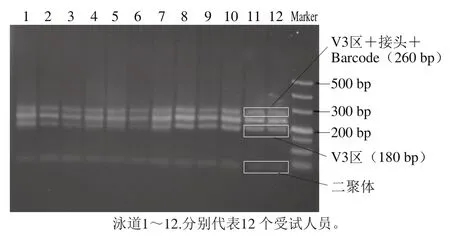
图1 肠道菌群16S rRNA V3区PCR扩增产物Fig.1 PCR fi ngerprints of the V3 region of the 16S rRNA of gut bacteria
图1为肠道菌群16S rRNA V3区PCR扩增产物建库后加上接头和Barcode的3%琼脂糖凝胶电泳图。最终获得的V3区测序文库大小集中在260 bp左右,完全符合细菌16S rRNA基因V3区平均长度为180 bp的要求,即测序长度可以覆盖V3区全长。
2.2 服用微生态制剂后肠道微生物α多样性的分析
2.2.1 多样性指数分析
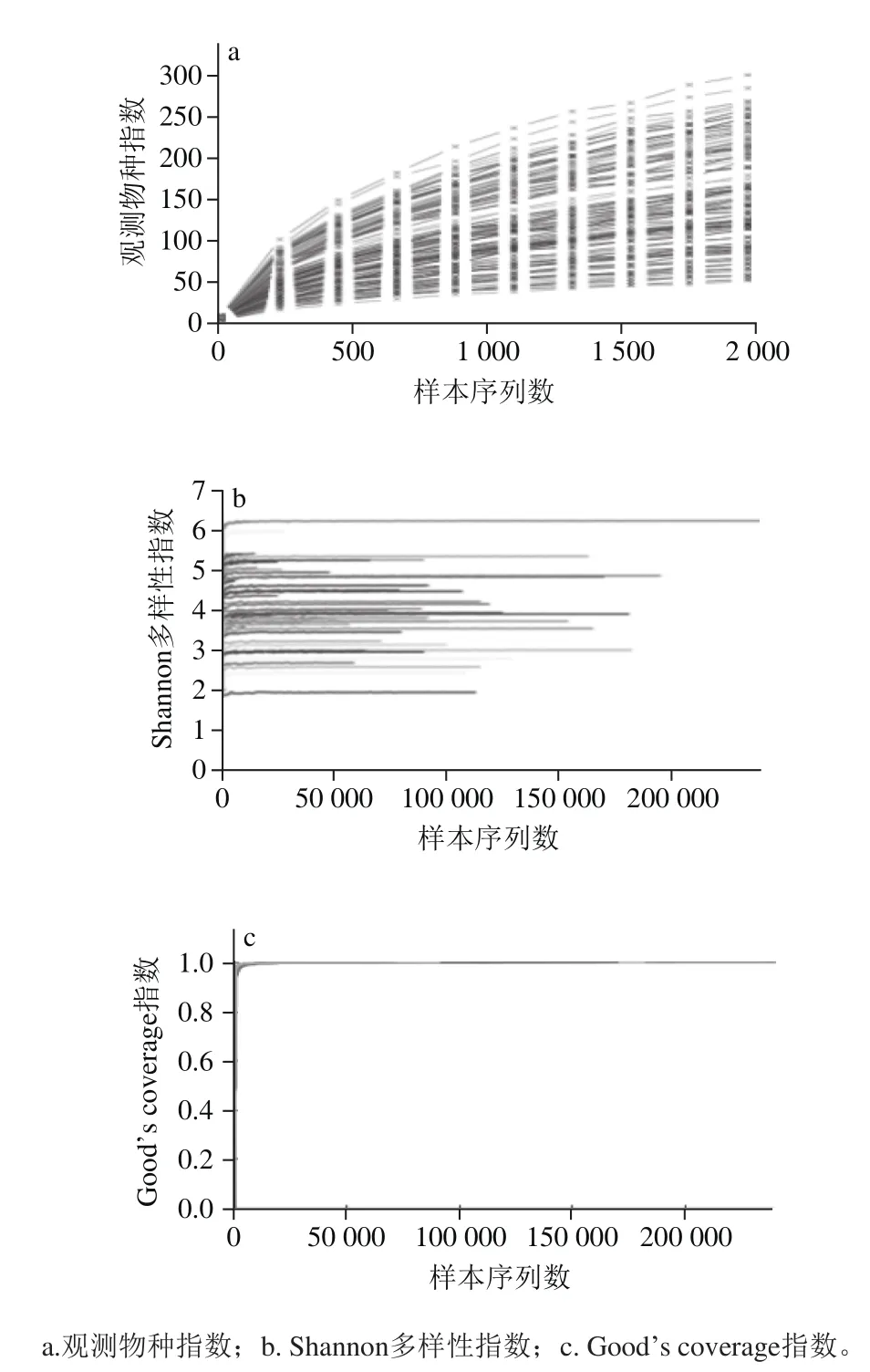
图2 样本测序分析Fig.2 Sequencing analysis of all samples
从图2中反映样本物种测序深度的观测物种指数、反映微生物多样性和均匀度的Shannon多样性指数以及反映测序覆盖率的Good’s coverage指数可以看出,各组样本的OTU数目随着测序深度的增加基本达到饱和,具有很高的测序覆盖率,说明当前测序深度足以发现各样本生境中的大部分物种,这与2.1节的结果一致。
2.2.2 微生态制剂干预下的肠道菌群多样性的变化
为研究微生态制剂对受试人群肠道微生物多样性的影响,从各样本肠道菌群的丰富度和均匀度的层面研究肠道微生物的多样性。首先通过反映样本物种丰富度的Ace指数来研究几组受试人群之间菌群多样性的差异,其次通过反映微生物多样性和均匀度的Shannon多样性指数来研究几组样本的多样性和均匀度的差异,发现微生态制剂对肠道微生物α多样性造成了一定程度的影响,结果如图3所示。
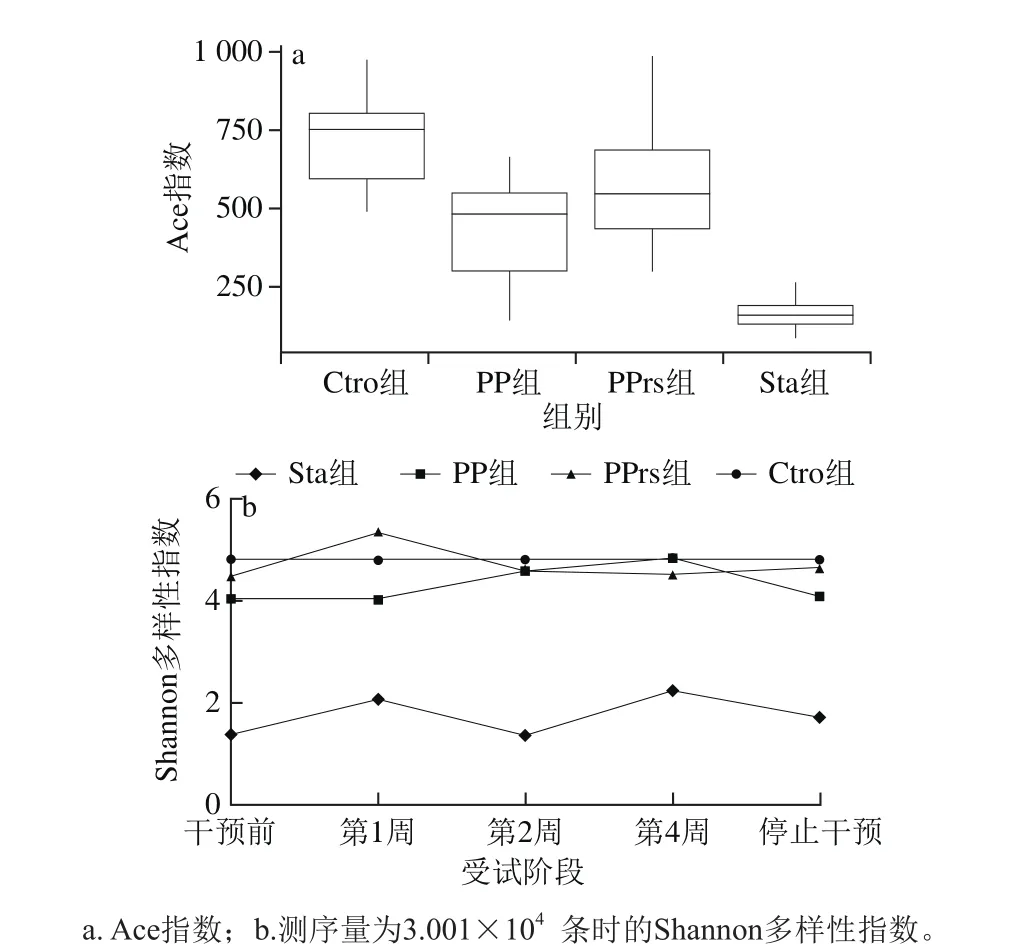
图3 微生态制剂对肠道微生物α多样性指数的影响Fig.3 Effect of microecologics on α diversity index of gut microbiota
图3a表示服用不同微生态制剂后志愿受试人群肠道菌群的Ace指数的变化,Ace指数反映了各组受试人群肠道菌群的物种丰富度,从中可看出3 种微生态制剂对肠道菌群的影响具有一定差异,其中,Sta和PP组受试人群肠道菌群丰度远不及PPrs组,说明PPrs对肠道菌群的多样性调节作用更明显。图3b结果表明PPrs组受试人群服用第1周后,肠道菌群的生物群落结构多样性明显好转,随着服用时间的延长,逐渐与Ctro组接近,而且在停止服用后,肠道生物群落结构多样性也没有大幅度降低,说明PPrs对调节肠道菌群结构具有良好的作用。与PPrs不同的是,服用PP的受试人群的肠道菌群的生物群落结构多样性在第1周没有明显好转,随着服用时间的延长,向Ctro组接近,并且停止服用后,肠道菌群的生物群落结构多样性恢复了原有状态。而Sta组受试人群肠道菌群的群落结构多样性明显低于其他组,但在服用Sta期间,肠道菌群的生物群落结构多样性有升高的趋势,而且在停止服用Sta后,肠道菌群的生物群落结构多样性较服用前有所升高,说明Sta也具有调整肠道菌群结构的作用。总体而言,PPrs的干预效果较其他两种制剂好,这也与图3a结果一致。
2.3 微生态制剂对肠道菌群整体结构的影响
2.3.1 肠道菌群结构分析
根据各变量之间的进化距离和丰度信息,基于加权UniFrac选取不同微生态制剂干预下的各组受试人群肠道菌群在属水平上的相对丰度的差异做PCoA分析,并根据分组的信息得到图4。
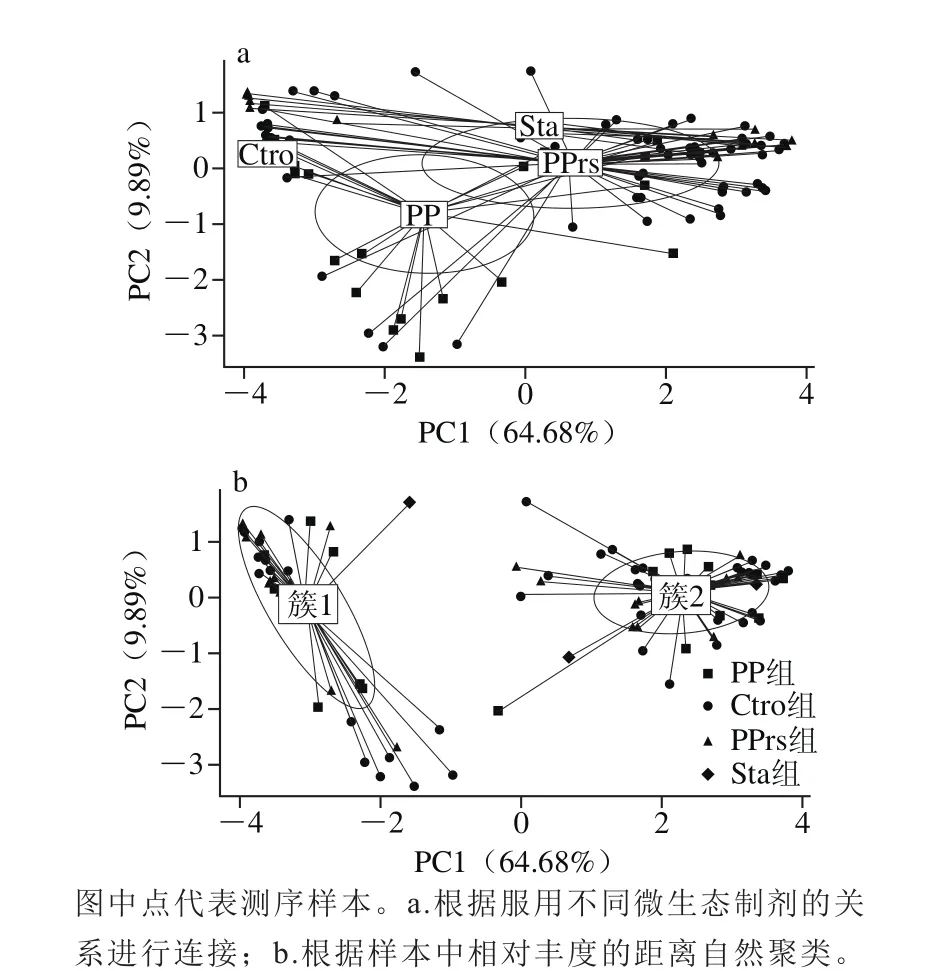
图4 基于加权UniFrac PcoA分析Fig.4 UniFrac weighted PcoA analysis
图4a中根据服用不同微生态制剂的关系进行连接,发现服用微生态制剂组样本与Ctro组有交叠的趋势,各组样本并不能完全分开,说明相比较其他因素,微生态制剂对肠道菌群结构的调节作用更为明显。在图4b中,根据样本中相对丰度的距离自然聚类为两簇,沿第一主成分也是贡献率最大的主成分(64.68%)完全分离,根据数据提示这与Prevotella和Bacteroides的相对丰度相关。此外,从图4a中可看出,PP组样本在X轴上距离较大,说明在PP干预下肠道菌群结构多样性变化较大;PPrs组人群的肠道菌群结构逐渐向Ctro组靠近,提示PPrs具有更强的改变肠道菌群失调的能力。
2.3.2 肠道菌群中不同分类水平相对丰度的分析
在QIIME中,通过Usearch在97%相似水平划分得到1 619 个OTUs,所有OTUs被划分进化水平,结果如图5所示。
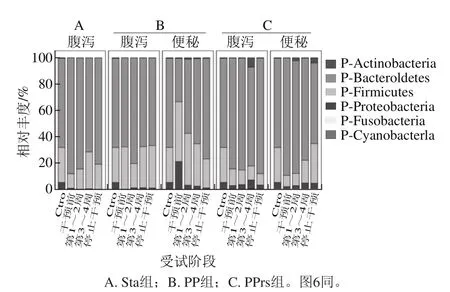
图5 益生菌制剂对人群肠道菌群门水平上相对丰度的影响Fig.5 Effect of microecologics on relative abundance of dominant intestinal bacterial phyla in human
在门的水平上,主要有放线菌门(Actinobacteria)、拟杆菌门(Bacteroidetes)、硬壁菌门(Firmicutes)、变形菌门(Proteobacteria)、梭杆菌门(Fusobacteria)及蓝藻细菌门(Cyanobacteria),其中绝大多数序列属于Bacteroidetes和Firmicutes,约占总序列数的94.37%。Sta组的肠道菌群结构中Firmicutes相对丰度有一定的增长,由14.08%升至22.4%,而Bacteroidetes和Proteobacteria相对丰度下降明显。PP组便秘人群的肠道菌群结构中Bacteroidetes相对丰度明显增加,而Firmicute、Proteobacterias和Actinobacteria都相应减少;而腹泻人群的Bacteroidetes相对丰度明显减少,只有Firmicutes明显增加。PPrs组腹泻人群在干预期间肠道菌群结构中除Firmicutes有较明显的减少外,Actinobacteria、Bacteroidetes和Proteobacterias都相应有所增长;而便秘组除Bacteroidetes外,Actinobacteria、Proteobacterias和Firmicutes都有明显有增长。
另外,通过Usearch在97%相似水平划分得到OTUs被划为科和属的水平共114 个,结果如图6所示。
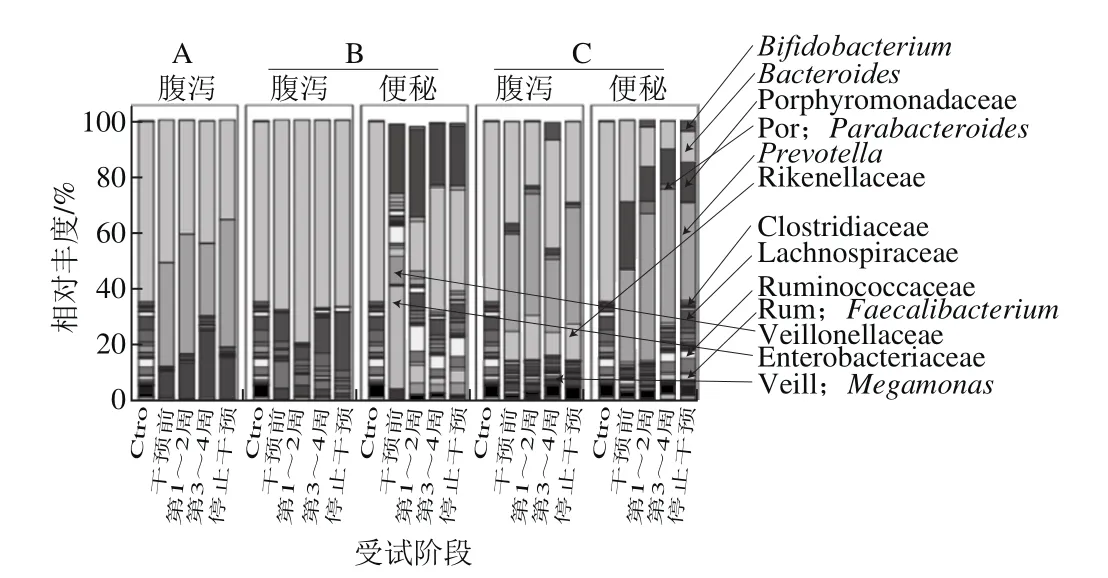
图6 微生态制剂对人群肠道菌群科和属水平上相对丰度的影响Fig.6 Effect of microecologics on relative abundance of intestinal bacterial genera
从整体结构中可以看出不同受试人群肠道菌群结构中科和属水平的差异较明显,服用微生态制剂后,属的相对丰度明显增加。健康受试人群的肠道菌群以Bacteroidales为核心菌属,而便秘和腹泻受试人群则以Bacteroidales、Prevotellaceae和Ruminococcaceae为主。就腹泻受试人群的肠道菌群而言,服用Sta和PPrs后,Bacteroidales和Prevotellaceae占相当大的比例,而服用PP后的Bacteroidales所占比重较大。便秘受试人群的肠道中以Prevotellaceae和Ruminococcaceae为主,服用PP后Bacteroidales的相对丰度降低,而服用PPrs的受试人群肠道菌群中这3 种菌属似乎处于一个相对平衡的状态,这需要进一步去挖掘;此外,Bifidobacterium、Lactobacillus、Parabacteroides这些常见的外源性益生菌均在受试者肠道菌群有增长迹象。

图7 肠道菌群丰度分布热图Fig.7 Heat map of relative abundance of gut microbiota
为了更好地体现微生态制剂作用下的肠道菌群相对丰度的分布,在属水平做了菌属丰度的分布热图,并进行了聚类,结果如图7所示。对比Sta组、PP组和PPrs组受试人群的肠道菌群,肠道中与产SCFAs相关的菌属都属于相对丰度较高的菌属,多属于Ruminococcaceae、Lachnospiraceae、Lactobacillaceae、Clostridiaceae、Bacteroidetes、Prevotellaceae、Enterobacteriaceae、Desulfovibrionaceae和Rikenellaceae。此外,PPrs中的Bif i dobacteriaceae、Lactobacillus和Parabacteroides有益菌虽然占有的丰度不高,但是在服用微生态制剂后明显有增长的趋势,这与2.3.1节得出的结果一致。在受试人群的肠道菌群中Citrobacter等可能性致病菌被检测出,随着服用微生态制剂时间的延长,其相对丰度降低。为进一步得到更多的信息,还需从中选择差异显著的菌群进行具体分析。
2.4 肠道菌群结构中差异显著的关键菌属鉴定
2.4.1 肠型分析
Wu等[23]按照肠道菌群的种类和丰度对样本进行肠型的归类,Prevotella和Bacteroides在健康成年人群的肠道菌群结构中占主导地位,与其他细菌一同为人体提供营养并维持肠道的正常生理,参与体内的拮抗反应,调节细菌毒素的产生,增强宿主固有的免疫反应。
结合肠型分析(图8):在干预前,健康人群肠道菌群内的Bacteroides相对丰度较高且稳定,便秘和腹泻人群肠道菌群中Bacteroides的相对丰度比Prevotella高。经过服用微生态制剂后,Sta组和PPrs组腹泻人群中的Bacteroides随时间呈下降趋势,相对丰度由高于50%降到低于20%,相应的Prevotella随时间呈上升趋势,肠型由Bacteroides型转换到Prevotella型;而便秘人群的Prevotella和Bacteroides的相对丰度会随着服用微生态制剂有所改变,变化比例没有使肠型改变。
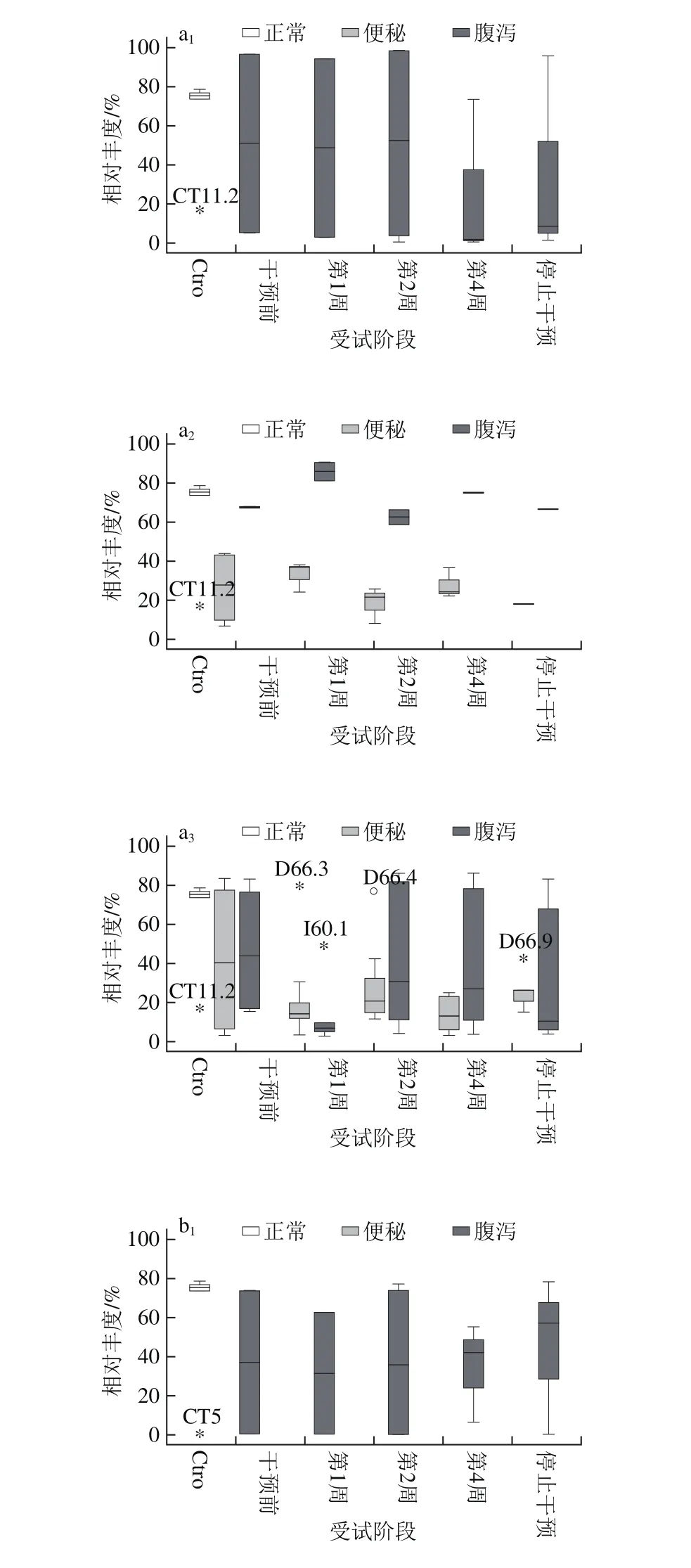
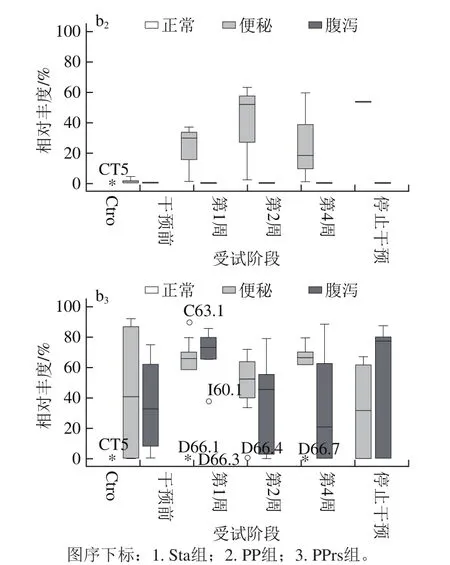
图8 微生态制剂对肠道中Bacteroides(a)和Prevotella (b)相对丰度的影响Fig.8 Effect of probiotics on relative abundance of Bacteroides (a) and Prevotella (b) in the intestine
2.4.2 关键菌属鉴定
根据肠道微生物整体结构的分析发现,微生态制剂干预前后的肠道微生物具有一定程度的差异,进一步在属水平数据上进行t检验显著性差异,发现服用Sta组受试人群的肠道结构有30 个属显著变化,PP组受试人群的肠道结构有41 个属显著变化,PPrs组受试人群的肠道结构约有68 个属显著变化,又通过线性判别分析的LEfSe差异分析算法实现降维评估差异物种的影响,结果如图9所示。总体而言,PPrs对肠道结构影响较明显。Sta组、PP组和PPrs组3 组受试人群的肠道菌群与产SCFAs相关的科、属都有明显增长,相较其他两种制剂,PP组受试人群产SCFAs相关菌属的相对丰度增长明显,尤其是Lachnospiraceae中的Blautia、Lachnospira以及Ruminococcaceae中的Faecalibacterium、Oscillospir等,其中Blautia和Faecalibacterium与SCFAs含量呈显著正相关。
此外,我们应用环境因素对数据进行约束,采用冗余分析(redundancy analysis,RDA)的方法,以健康、便秘与腹泻分组作为起约束作用的解释变量,用于预测和解释全部肠道菌属的相对丰度数据组成的响应变量,结果如图10所示,相对丰度数据中约有14%的变异度能够被健康、便秘与腹泻环境变量所解释,能够代表便秘和腹泻与健康人群肠道菌群结构差异显著相关的关键菌。
在RDA排序图中可以看到,位于3 种状态下的菌均有显著不同,腹泻状态下的肠道菌群多样性要明显低于便秘和健康状态。其中健康状态丰度较高的菌属多属于Lachnospiraceae、Veillonellaceae、Streptococcaceae、Erysipelotrichaceae等产SCFAs细菌。腹泻状态下Megamonas的丰度较明显,可能属于腹泻的关键菌,有待进一步探究。便秘状态下肠道菌群处于较复杂的状态,其中Ruminococcaceae、Rikenellaceae、S24-7、Clostridiales、Prevotella以及Lachnospiraceae、Veillonellaceae、Erysipelotrichaceae等产SCFAs菌数量较多。
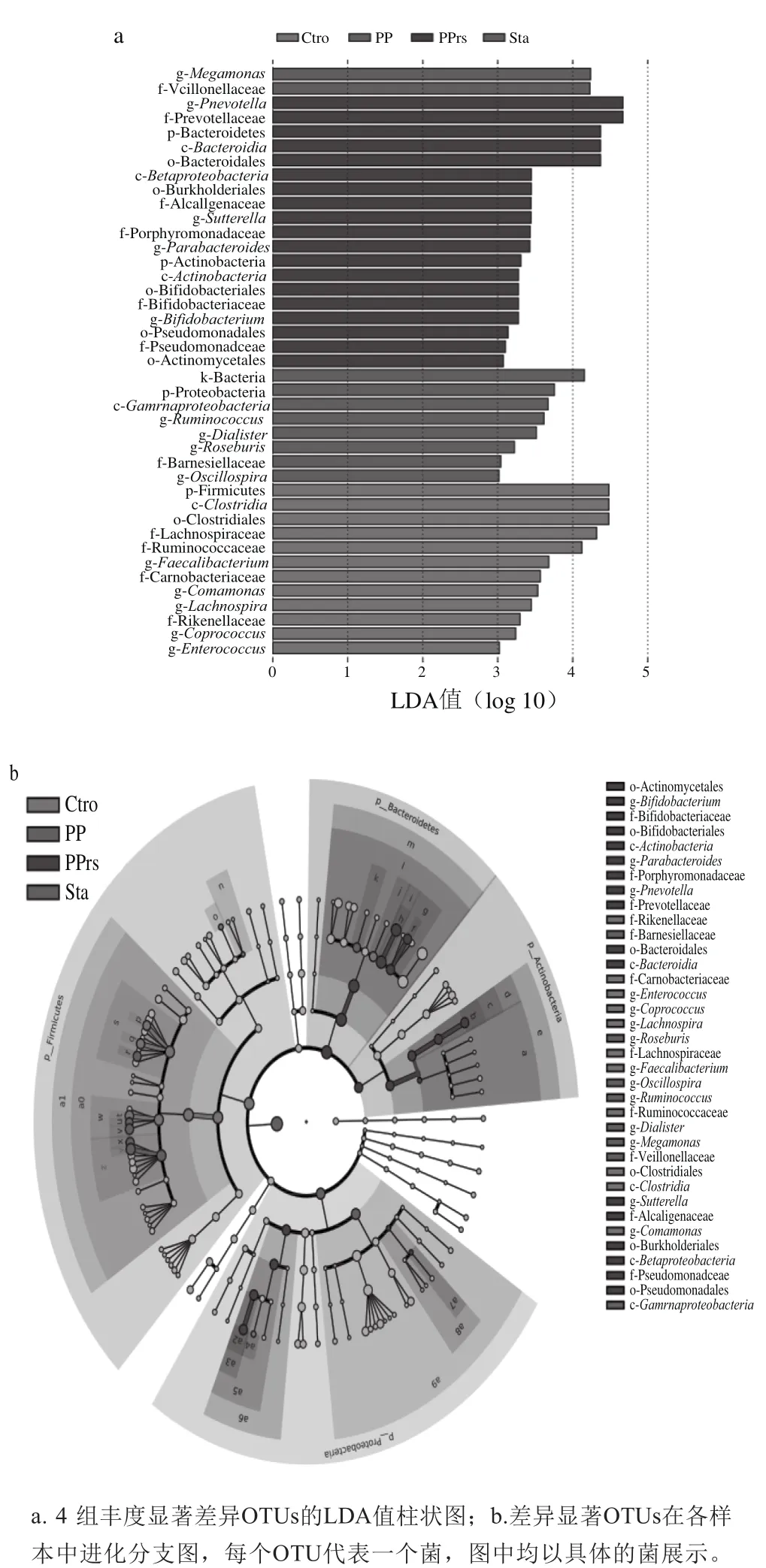
图9 肠道菌群中关键菌属的变化Fig.9 Changes in key bacterial genera in the intestinal fl ora
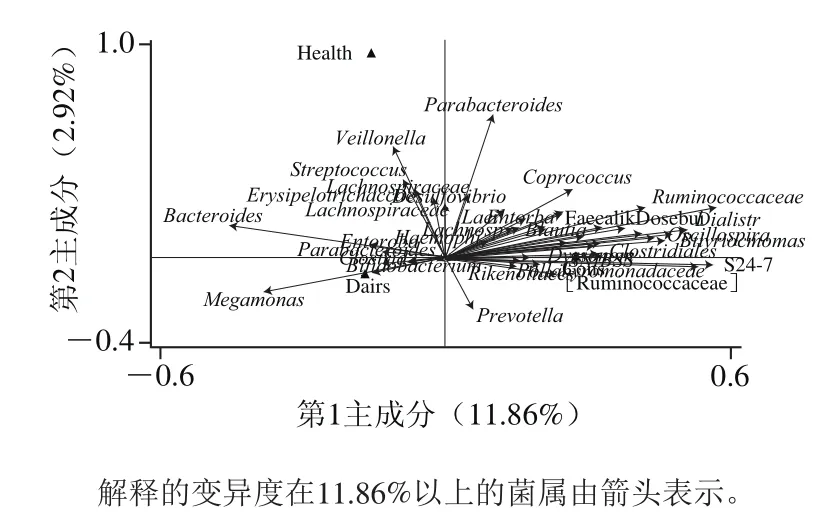
图10 冗余分析图Fig.10 Biplot of redundancy analysis
2.5 微生态制剂对粪便中SCFAs表达水平的影响
为对肠道菌群特异性菌属及其代谢产物进一步分析,用气相色谱法检测粪便样本中SCFAs的含量,从表1可看出,在测定范围内各样本均具有较好的线性关系,相关系数均达到0.998以上。

表1 SCFAs的回归方程及相关系数Table1 Regression equations with correlation coefficients for SCFAs
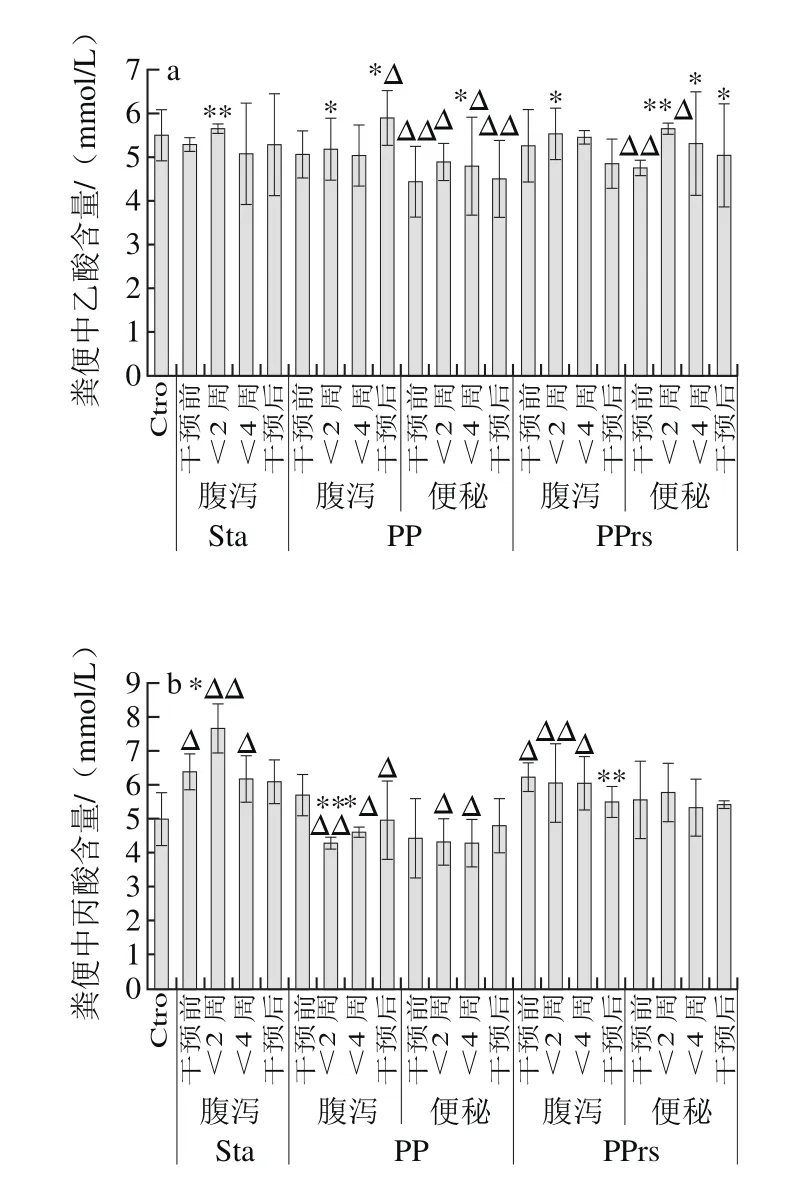
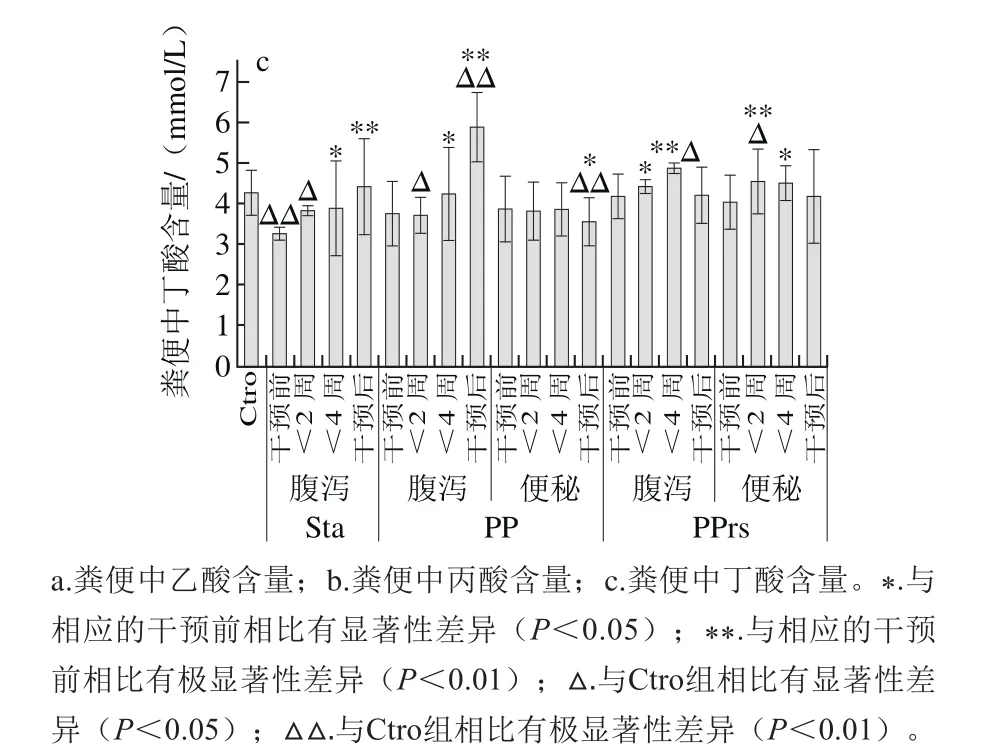
图11 服用微生态制剂后对粪便中SCFAs含量的影响Fig.11 Effect of microecologics on SCFAs in feces
肠道菌群微生态制剂干预后,粪便中检测的SCFAs多为乙酸、丁酸和丙酸,这些SCFAs都能在短时间内被结肠细胞吸收。在Sta组、PP组和PPrs组受试人群的肠道菌群中,与产SCFAs相关的菌属都有明显的增长,其中PP组中产SCFAs相关菌属的相对丰度要比其他两组增长明显。
如图11所示,与Ctro组相比,服用不同微生态制剂后,SCFAs含量存在显著性差异。服用Sta后,丙酸含量显著增加,乙酸与丁酸也在2周左右有所增加,随着服用时间延长尤其是停止服用后,SCFAs的含量基本有所增加。腹泻人群肠道菌群结构单一,服用PPrs后肠道菌群多样性增高。受试人群在服用PP期间,不论是便秘还是腹泻状态,只有乙酸的含量增加,丙酸和丁酸甚至呈现降低的现象。尤其腹泻人群在停止服用后SCFAs含量有着显著差异。服用PP的受试人群肠道中Veillonellaceae、Ruminococcaceae、Lachnospiraceae都增长较快,可能与服用的微生态制剂内包含的菌种有关。受试人群在服用PPrs后,乙酸和丁酸含量都在明显增加,而且停止服用后,SCFAs的含量与正常对照组也没有显著差异。
3 讨 论
近年来,很多研究表明肠道菌群与宿主的健康状况息息相关,密切影响着宿主的营养吸收、物质代谢、免疫以及胃肠道发育等各个方面,宿主自身的基因型、年龄和免疫系统等因素在肠道菌群结构的形成和稳定中起到了重要作用[24]。微生态制剂作为一种无毒、无污染的环保产品对维持肠道菌群与宿主稳态方面起着重要的作用。
本研究中,服用3 种微生态制剂前后肠道菌群的多样性及丰度都有着不同程度的变化,并且,服用微生态制剂组与对照组有交叠的趋势,疾病状态下的受试人群肠道菌群结构在微生态制剂的调节下都不同程度地朝着健康状态发展,说明微生态制剂具有调节肠道菌群结构的作用,这与张家超等[25]的研究结论一致。在门的水平上,测得主要有放线菌门(Actinobacteria)、拟杆菌门(Bacteroidetes)、硬壁菌门(Firmicutes)、变形菌门(Proteobacteria)、梭杆菌门(Fusobacteria)及蓝藻细菌门(Cyanobacteria),其中绝大多数序列属于Bacteroidetes和Firmicutes,约占总序列数的94.37%,这与前人的研究结果[26]相似,它们对机体健康起着关键作用。3 种微生态制剂中,服用Sta的受试人群肠道菌群丰度依然较低,但是较干预前还是有一定的积极调控作用,在Sta的作用下Firmicutes增加,这可能与腹泻状态人群本身菌群多样性少有关;PP的作用效果具有时效性,对肠道菌群中Bacteroidetes和Firmicutes有明显影响,减少了便秘人群肠道中Firmicute、Proteobacterias和Actinobacteria,并使Bacteroidetes相对丰度明显增加,而只增加了腹泻人群肠道中的Firmicutes,这些现象只发生在服用期间,停止服用后又恢复了原有状态;PPrs作用效果最好,不仅仅具有时效性,而且在停止服用后肠道菌群不会恢复到原有的疾病状态,腹泻组除了Firmicutes有比较明显的减少外,相应Actinobacteria、Bacteroidetes和Proteobacterias都有明显的增长;而便秘组除Bacteroidetes外,Actinobacteria、Proteobacterias和Firmicutes有明显的增长。说明复合制剂的作用效果优于单一制剂,这与曲巍等[27]得出的结论一致,摄入适量的复合微生态制剂对调节肠道菌群的平衡具有一定的帮助,相应菌门的消长与微生态制剂的成分有很大关系,并且,肠道菌群的多样性还与受试人群的健康状态有关,比如,便秘人群较腹泻人群的多样性要高。
另外,在属水平上,本研究划分所得的114 个属中有近30 个菌属丰度总和达到肠道菌群的90%以上。不同受试人群肠道内核心菌属呈现较大的差异性,主要分为Bacteroides和Prevotella两种肠型,这与Arumugam等[28]划分结果一致,也就是说,3 种微生态制剂对于核心菌属的改变几乎无影响,说明相较于短期的微生态制剂干预,长期固定的膳食结构对肠道菌群结构的形成及稳定有着深远的影响。此外,在微生态制剂干预下,Lachnospiraceae中的Blautia、Lachnospira以及Ruminococcaceae中Faecalibacterium、Oscillospir等与产SCFAs相关的菌属都有明显的增长,其中Blautia和Faecalibacterium与SCFAs含量呈显著正相关,并且PP组产SCFAs相关菌属的相对丰度最高,而PPrs组中Bifidobacterium、Lactobacillus、Butyricimonas、Parabacteroides这些常见的外源性益生菌都有明显的增长,并且,随着服用微生态制剂时间的延长,可能性致病菌相对丰度降低,提示微生态制剂中的益生菌在促进产SCFAs相关菌增加并升高SCFAs含量的同时,也能抑制有害菌的增长,这与益生菌的生理功能[29]密不可分,这也是微生态制剂有益于人体肠道健康,特别是对肠道炎症以及结直肠癌均具有治疗作用[30]的重要原因。
多项研究表明,SCFAs主要包括乙酸、丙酸、丁酸,并且,结肠厌氧菌发酵小肠未消化吸收的碳水化合物产生乙酸,丙酸为拟杆菌门发酵的主要产物,丁酸主要由厚壁菌门代谢产生[31-33],肠道中产SCFAs的相关菌属及其代谢产生的SCFAs对宿主肠屏障功能的维护有重要作用[17]。本研究中,服用不同微生态制剂后,粪便中SCFAs含量存在显著性差异,这与个体及采集样本时构成的差异等因素相关。由于Sta在体内能完整地进入结肠而不会被人体消化酶水解,可作为双歧因子被双歧杆菌利用后产生大量的SCFAs,服用Sta后,丙酸、乙酸与丁酸这3 种SCFAs的含量较干预前都有所增加,这与赵杰等[34]得出的结论相似,即Sta给肠道菌群提供发酵底物,产SCFAs的菌种能快速而且大量增长,也不会随着外源性微生态制剂的消失快速死亡。服用PPrs的腹泻人群从服用到终止以后的一段时间,乙酸和丁酸含量都明显增加,SCFAs的含量与正常对照组没有显著差异;服用PP的人群,不论是便秘还是腹泻的状态,只有乙酸的含量有所增加,丙酸和丁酸甚至呈现降低的现象,尤其腹泻人群在停止服用后SCFAs含量有显著差异。此外,服用PP的受试人群肠道中Veillonellaceae、Ruminococcaceae、Lachnospiraceae都增长较快,该结果可能与服用的微生态制剂的组成成分有关,微生态制剂的4 种益生菌均有构成生物屏障、降低胆固醇并产生有益代谢产物的作用,如此看来,如果摄入PP,肠道菌群中关键菌属会快速增长,但是代谢产物含量不会太高。
4 结 论
本研究以受试人群健康状况作为选择的依据,探讨了不同微生态制剂干预后,受试人群肠道菌群结构的变化与产SCFAs相关菌属的相关性,应用Ion torrent PGM二代测序技术和多变量统计学分析方法,获得的结论与近些年来国内外研究成果具有一致性。研究显示受试人群尤其PP组和PPrs组中肠道菌群结构多样性随干预时间的延长而有明显增加,主要表现为Bacteroidetes和Firmicutes多样性显著增加,提示食用微生态制剂会改善肠道菌群结构的多样性,也提示肠道中相应菌属的消长均与微生态制剂的成分有关,即PPrs比PP和Sta的作用效果和调节能力更突出。研究还表明,微生态制剂干预后促进了肠道内产SCFAs菌属增长,提高了外源性益生菌的丰度和抑制了条件致病菌的增长。总之,肠道疾病状态下的人群服用微生态制剂后,具有向健康人群的肠道菌群结构调整的趋势,菌群多样性和SCFAs表达水平提高,有利于肠道菌群结构的稳态和健康。因此本研究结论为:微生态制剂有改变腹泻、便秘人群肠道菌群整体结构的功效,证明了与产SCFAs菌属的丰度提高有关,进一步明确了利用微生态制剂调节和恢复便秘、腹泻人群肠道健康的科学价值。
[1] QIN J, LI R, RAES J, et al. A human gut microbial gene catalogue established by metagenomic sequencing[J]. Nature, 2010, 464: 59-65.DOI:10.1038/nature08821.
[2] SINA C, GAVRILOVA O, FÖRSTER M, et al. G protein-coupled receptor 43 is essential for neutrophil recruitment during intestinal inf l ammation[J]. Journal of Immunology, 2009, 183(11): 7514-7522.DOI:10.4049/jimmunol.0900063.
[3] MORRISON D J, PRESTON T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism[J]. Gut Microbes, 2016, 7(3): 189-200. DOI:10.1080/19490976.2015.1134082.
[4] FLINT H J, DUNCAN S H, SCOTT K P, et al. Links between diet,gut microbiota composition and gut metabolism[J]. Proceedings of the Nutrition Society, 2015, 74(1): 13-22. DOI:10.1038/nrneph.2015.191.
[5] PETRA L, PAULINE Y, GRIETJE H, et al. Diversity of human colonic butyrate-producing bacteria revealed by analysis of the butyryl-CoA:acetate CoA-transferase gene[J]. Environmental Microbiology, 2010,12(2): 304-314. DOI:10.1111/j.1462-2920.2009.02066.
[6] LIOU A P, PAZIUK M, JESUS-MARIO L, et al. Conserved shifts in the gut microbiota due to gastric bypass reduce host weight and adiposity[J]. Science Translational Medicine, 2013, 5(178): 178-210.DOI:10.1126/scitranslmed.3005687.
[7] VANDEPUTTE D, FALONY G, VIEIRASILVA S, et al. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates[J]. Gut, 2015,65(1): 57-62. DOI:10.1136/gutjnl-2015-309618.
[8] FESTI D, SCHIUMERINI R, MARASCO G, et al. Gut microbiota and metabolic syndrome[J]. Internal & Emergency Medicine, 2013,8(Suppl 1): 11-15. DOI:10.1007/s11739-013-0916-z.
[9] SHEN Q, MAITIN V. Chapter 11: obesity-associated gut microbiota:characterization and dietary modulation[M]// TUOHY K, DEL RIO D.Diet-microbe interactions in the gut. NewYork: Academic Press, 2015:149-171. DOI:10.1016/B978-0-12-407825-3.
[10] MUGAMBI M N, MUSEKIWA A, LOMBARD M, et al. Synbiotics,probiotics or prebiotics in infant formula for full term infants:a systematic review[J]. Nutrition Journal, 2012, 11(7): 1-32.DOI:10.1186/1475-2891-11-81.
[11] DERRIEN M, VAN HYLCKAMA VLIEG J E. Fate, activity, and impact of ingested bacteria within the human gut microbiota[J].Trends in Microbiology, 2015, 23(6): 354-366. DOI:10.1016/j.tim.2015.03.002.
[12] ELOE-FADROSH E A, BRADY A, CRABTREE J, et al. Functional dynamics of the gut microbiome in elderly people during probiotic consumption[J]. Mbio, 2015, 6(2): 544-556. DOI:10.1128/mBio.00231-15.
[13] 杨远志, 李发财, 庞明利, 等. 益生菌和益生元在功能性食品中的应用现状及展望[J]. 中国食品添加剂, 2009(6): 187-192. DOI:10.3969/j.issn.1006-2513.2009.06.037.
[14] 郭壮. 应用焦磷酸测序技术对不同人群肠道微生物群落结构的研究[D]. 无锡: 江南大学, 2013: 59-73.
[15] LAURA R, RICHARD A, VIJAY S, et al. Quantitative profiling of gut microbiota of children with diarrhea-predominant irritable bowel syndrome[J]. American Journal of Gastroenterology, 2012, 107(11):1740-1751. DOI:10.1038/ajg.2012.287.
[16] 王友湘, 陈庆森. 瑞士乳杆菌对小鼠肠道微生物区系的影响[J]. 食品科学, 2008, 29(9): 542-546. DOI:10.3321/j.issn:1002-6630.2008.09.129.
[17] 贾彦, 魏秀秀, 赵培, 等. 魔芋葡甘露聚糖和香蕉抗性淀粉对小鼠肠道短链脂肪酸分泌及血脂影响的研究[J]. 现代食品科技, 2016,32(7): 18-25. DOI:10.13982/j.mfst.1673-9078.2016.7.004.
[18] 王水泉, 包艳, 董喜梅, 等. 植物乳杆菌的生理功能及应用[J]. 中国农业科技导报, 2010, 12(4): 49-55. DOI:10.3969/j.issn.1008-0864.2010.04.10.
[19] 郑红星, 祁珊珊. 嗜酸乳杆菌的研究进展[J]. 黑龙江畜牧兽医,2015(17): 61-64. DOI:10.13881/j.cnki.hljxmsy.2015.1561.
[20] 王英, 周剑忠, 仇小妹, 等. 益生菌干酪乳杆菌FM10-3生物学与发酵特性分析[J]. 江苏农业学报, 2013, 29(3): 654-658. DOI:10.3969/j.issn.1000-4440.2013.03.034.
[21] 韩俊燕. 双歧杆菌生理特性与其肠道定殖能力相关性的研究[D]. 无锡:江南大学, 2016: 22-24.
[22] CAPORASO J G, KUCZYNSKI J, STOMBAUGH J, et al. QIIME allows analysis of high-throughput community sequencing data[J].Nature Methods, 2010, 7(5): 335-336. DOI:10.1038/nmeth.f.303.
[23] WU G D, CHEN J, HOFFMANN C, et al. Linking long-term dietary patterns with gut microbial enterotypes[J]. Science, 2011, 334: 105-108. DOI:10.1126/science.1208344.
[24] ZHANG C H, ZHANG M H, PANG X Y, et al. Structural resilience of the gut microbiota in adult mice under high-fat dietary perturbations[J].Isme Journal, 2012, 6(10): 1848-1857. DOI:10.1038/ismej.2012.27.
[25] 张家超, 郭壮, 孙志宏, 等. 益生菌对肠道菌群的影响: 以Lactobacillus casei Zhang研究为例[J]. 中国食品学报, 2011, 11(9):58-62. DOI:10.16429/j.1009-7848.2011.09.001.
[26] BEZIRTZOGLOU E, TSIOTSIAS A, WELLING G W, et al.Microbiota profile in feces of breast- and formula-fed newborns by using fluorescence in situ hybridization (fish)[J]. Anaerobe, 2011,17(6): 478-482. DOI:10.1016/j.anaerobe.2011.03.009.
[27] 曲巍, 张智, 马建章, 等. 高通量测序研究益生菌对小鼠肠道菌群的影响[J]. 食品科学, 2017, 38(1): 214-219. DOI:10.7506/spkx1002-6630-201701036.
[28] ARUMUGAM M, RAES J, PELLETIER E, et al. Enterotypes of the human gut microbiome[J]. Nature, 2011, 506: 174-180. DOI:10.1038/nature09944.
[29] 温俊, 孙冬岩, 孙笑非. 肠道菌群的重要性及微生态制剂对肠道的调节作用[J]. 饲料研究, 2010(2): 70-72. DOI:10.13557/j.cnki.issn1002-2813.2010.02.018.
[30] 梁淑文, 屈昌民, 王晓英, 等. 益生菌治疗结肠癌患者术后化疗相关性腹泻的效果观察[J]. 现代生物医学进展, 2014, 14(24): 4686-4688.DOI:10.13241/j.cnki.pmb.2014.24.020.
[31] BIANCHI F, DALL’ASTA M, DEL RIO D, et al. Development of a headspace solid-phase microextraction gas chromatography-mass spectrometric method for the determination of short-chain fatty acids from intestinal fermentation[J]. Food Chemistry, 2011, 129(1): 200-205. DOI:10.1016/j.foodchem.2011.04.022.
[32] NAKAMURA N, HAMAZAKI T, JOKAJI H, et al. Effect of HMGCoA reductase inhibitors on plasma polyunsaturated fatty acid concentrations in patients with hyperlipidemia[J]. International Journal of Clinical & Laboratory Research, 1998, 28(3): 192-195.DOI:10.1007/s005990050043.
[33] PRYDE S E, DUNCAN S H, HOLD G L, et al. The microbiology of butyrate formation in the human colon[J]. FEMS Microbiology Letters,2002, 217(2): 133-139. DOI:10.1016/S0378-1097(02)01106-0.
[34] 赵杰, 朱维铭, 李宁. 益生菌、益生元、合生元与炎症性肠病[J].肠外与肠内营养, 2014, 21(4): 251-253; 256. DOI:10.16151/j.1007-810x.2014.04.014.
