湖北地区一扩张型心肌病家系致病基因筛查
2017-12-19吕永楠徐红新王京伟张宗玮
吕永楠,徐红新,王京伟,张宗玮
(武汉大学人民医院心内科1、检验科2,湖北 武汉 430060)
湖北地区一扩张型心肌病家系致病基因筛查
吕永楠1,徐红新1,王京伟2,张宗玮1
(武汉大学人民医院心内科1、检验科2,湖北 武汉 430060)
目的对湖北地区一个扩张型心肌病家系成员进行致病候选基因筛查,寻求家族性扩张型心肌病致病基因,探讨基因型和表型关系。方法先证者及其家族成员来自湖北省大冶市,先证者于2017年4月在武汉大学人民医院确诊为扩张型心肌病,已有家族成员死亡。详细询问先证者及其家属成员病史、家族史,并进行体格检查、血液指标、心脏超声和心电图检查。对患者的病史、家族史及检查结果分析。与先证者及其家属签订知情同意书,由武汉大学人民医院临床分子诊断中心对先证者候选致病基因全外显子高通量测序,获得可疑突变后,利用Sanger测序验证家系成员是否存在可疑突变。结果家系先证者(Ⅲ3)和妹妹(Ⅲ2)携带肌联蛋白(TTN)c.100126A>G(P.Thr33376Ala)错义突变。先证者目前心功能下降并伴有恶性心律失常,而其妹妹无明显临床症状,心脏超声检查无异常。结论本研究发现湖北地区一家族性扩张型心肌病家系存在TTN基因c.100126A>G(p.Thr33376Ala)错义突变,TTN与扩张型心肌病密切相关,是家族性扩张型心肌病重要致病基因。
扩张型心肌病;基因突变;肌联蛋白
扩张型心肌病(dilated cardiomyopathy,DCM)是一种以左室或双心室扩张伴有收缩功能损害为特点的心肌病,主要临床症状表现为心力衰竭、心律失常和栓塞性中风,更严重的是心源性猝死,5年内患者病死率达15%~50%[1]。研究发现约30%~50%扩张型心肌病患者具有家族性遗传,称为家族性扩张型心肌病(familial dilated cardiomyopathy,FDCM)[2]。患者发病年龄在20~30岁之间,病情进展快,生活质量差和预后不佳,死亡率高。目前除了心脏移植没有彻底的治疗办法,早期诊断和干预是延缓患者病情进展的有效手段。家族性扩张型心肌病显著的遗传特征,主要由基因突变所致,其最大的遗传特征为临床遗传异质性。已明确与家族性扩张型心肌病相关的致病基因约为65个,遗传模式主要表现为常染色体显性遗传[3]。本研究对一个扩张型心肌病家系成员进行了基因筛查,为早期筛查和干预扩张型心肌病提供候选基因。
1 资料与方法
1.1 研究对象 先证者及其家族成员来自湖北省大冶市,先证者于2017年4月在武汉大学人民医院确诊为扩张型心肌病,已有家族成员死亡。
1.2 临床资料 详细询问先证者及其家属成员病史、家族史,根据患者描述,制作患者家系系谱图(见图1),对先证者进行体格检查、血液指标、心脏超声和心电图检查等检查,对家族成员行心脏超声检查。
1.3 基因筛查 本研究与先证者及其家属签订知情同意书。由武汉大学人民医院临床分子诊断中心对先证者候选致病基因全外显子高通量测序,获得可疑突变后,利用Sanger测序验证家系成员是否存在可疑突变。
2 结 果
2.1 一般临床情况 患者,男,46岁。近一周上楼或活动量大时感胸闷、喘气不适,伴乏力,经休息后可减轻,不伴咳嗽、咳血,无胸痛、头昏等不适,未予以特殊处理,近2 d来患者感上述症状加重,活动耐量下降,伴夜间不能平卧入睡。查体:血压(BP)130/80 mmHg(1 mmHg=0.133 kPa),呼吸(R)26次/min,心率(P)88次/min,体温(T)36.3℃,神清,精神可,全身皮肤黏膜未见出血点、黄染,全身浅表淋巴结未及肿大,颈静脉无怒张,颈软,双肺呼吸音清,左下肺可闻及细湿啰音,心界向左下扩大,P 88次/min,律齐,未闻及明显杂音。腹平软,无压痛及反跳痛,肝脾肋下未及,双肾区无叩痛,双下肢无水肿。辅助检查:心脏超声—左房内径(LAD)=50 mm,左心室舒张期内径(LVDd)=62 mm,右房内径(RAD)=41 mm,右心室直径(RVD)=21 mm,左室射血分数(LVEF%)=35%。长程心电图:窦性心律,平均心率72次/min,室性早搏2 357个,短阵室速32阵。N端前脑钠肽(NT-proBNP)8 734 ng/L。
2.2 发病家族成员情况 Ⅰ1和Ⅰ2死亡原因不明,Ⅱ1和Ⅱ2诊断为扩张型心肌病,已死亡。Ⅲ5是先证者弟弟,于26岁死亡。Ⅲ7是先证者姐姐,于33岁死亡。Ⅲ2是先证者妹妹,年龄43岁,目前未发病。心脏超声检:LAD=26 mm,LVDd=41 mm,RAD=31 mm,RVD=18 mm,LVEF%=60%。Ⅳ1~Ⅳ7目前未发病,心脏超声检查无明显异常(见图1)。

图1 扩张型心肌病患者家系系谱图
2.3 候选基因筛查结果 实验室对受检者外周血提取基因组DNA采用Life Technologies公司Ion PGM高通量测序仪检测ACTC1、ACTN2等65个扩张性心肌病病相关基因,并使用Ion Reporter数据分析软件对测序结果进行分析,受检者共发现583个变异,通过分析ClinVar数据库、UCSC数据库Common SNPs、dbSNP 数据库及 1000-genome-project MAF≥1%的常见突变,滤除无功能的同义突变和内含子突变,得到候选有害或可能有害突变5个(见表1)。通过SIFT和PROVEAN预测变异的有害性,其中c.100126A>G(p.Thr33376Ala)变 异 为 Del eterious/Damaging,c.49762G>A(p.Val16588Met)变异为 Neutral/Damaging;c.17456G>A(p.Arg5819Gln)变 异 为Neutral/Tolerated;c.15758C>T(p.Thr5253Ile)变 异 为Deleterious/Tolerated。根据已知的家族发病史,初步认定c.100126A>G(p.Thr33376Ala)变异可能为致病变异,该位点致病可能表现常染色体显性遗传(见图2)。

图2 Sanger法序列验证先证者结果

表1 先证者基因突变结果
2.4 家系成员Sanger测序结果 根据先证者候选基因筛查结果,对获取的家族人员血液标本进行c.100126A>G(p.Thr33376Ala)基因Sanger测序,结果提示Ⅲ2是突变基因携带者(见图3)。
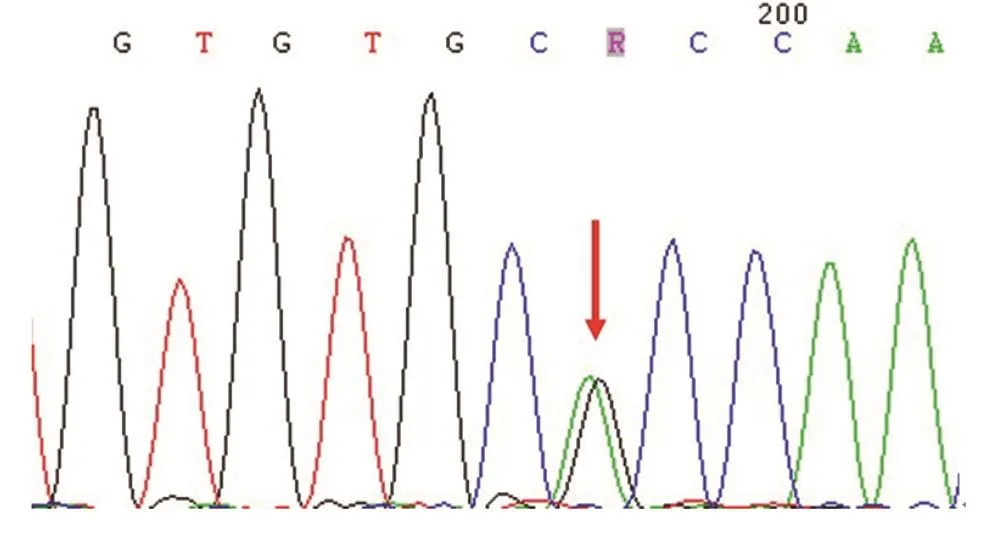
图3 Sanger法序列验证Ⅲ2结果
3 讨 论
扩张型心肌病的遗传模式主要表现为常染色体显性遗传,但少部分也存在其他遗传模式:常染色体隐性遗传、X连锁遗传和线粒体DNA遗传。本研究检测涵盖扩张型心肌病发病相关的致病基因有65个,但约75%的扩张型心肌病患者都集中在以下几个基因上:肌联蛋白(TTN)、心脏β-肌球蛋白重链(MYH7)、心肌肌球蛋白结合蛋白C(MYBPC3)、核纤层蛋白A/C(LMNA)、电压门控-钠通道α亚基(SCN5A)、心脏肌钙蛋白T 2(TNNT2)、心脏肌钙蛋白I 3(TNNI3)、心脏α-肌球蛋白重链(MYH6)、mtDNA和受磷蛋白(PLN)[4-6]。
本研究发现这一扩张型心肌病家系存在TTN基因突变,突变位点为c.100126A>G(p.Thr33376Ala)。TTN编码巨蛋白肌联蛋白,肌联蛋白是肌小节的重要组成单元,由大约33 000个氨基酸组成,在心脏功能中起着重要作用[7]。目前研究发现,25%的家族性扩张型心肌病是由TTN基因突变引起,TTN是扩张型心肌病的主要致病基因[8]。25%的扩张型心肌病患者可通过二代基因检测技术发现TTN截断突变,特别是杂合子的TTN截断突变已经被报道是扩张型心肌病的最常见遗传因素[9]。此外,Herman等[8]还发现TTN突变还包括错义突变、无义突变、框移突变和重复数突变,这些突变都可以改变氨基酸序列,并将他们归类为TTN截断突变。通过对湖北地区这一扩张型心肌病家系基因筛查研究结果分析,发现TTN基因c.100126A>G(p.Thr33376Ala)位点突变为错义突变,先证者和Ⅲ2均是此基因携带者,先证者已发病,但Ⅲ2目前无症状及心脏超声检查结果无异常,可能为遗传异质性导致,修饰基因可能在其中起到了重要作用。
遗传因素在家族性扩张型心肌病病理发展过程中起到了重要的作用,从基因水平寻找家族性扩张型心肌病发病机制具有意义,能够早期辅助临床诊断疾病,并预测患者疾病风险,制定个体化治疗方案,将为家族性扩张型心肌病的诊断和治疗开辟新的途径。但是临床实践中,基因检测仍然面临着许多问题和挑战,例如基因型和表型之间的差异,但是随着新一代基因检测技术的发展,分子遗传的研究将有助于明确基因型和表型之间的关系。
[1]Komajda M,Jais JP,Reeves F,et al.Factors predicting mortality in idiopathic dilated cardiomyopathy[J].Eutopean Heart Journal,1990,11(9):824-831.
[2]Mcnally EM,Golbus JR,Puckelwartz MJ.Genetic mutations and mechanisms in dilated cardiomyopathy[J].J Clin Invest,2013,123(1):19-26.
[3]Hershberger RE,Morales A,Sicgfried JD.Clinical and genetic issues in dilated cardiomyopathy:a review for genetics professionals[J].Genet Med,2010,12(11):655-667.
[4]Hampel H,Bennett RL,Buchanan A,et al.A practice guideline from the American college of medical genetics and genomics and the National society of genetic counselors:referral indications for cancer predisposition assessment[J].Genet Med,2015,17(1):70-87.
[5]Fatkin D.Guidelines for the diagnosis and management of familial dilated cardiomyopathy[J].Heart Lung Circ,2011,20(11):691-693.
[6]Hershberger RE,Siegfried JD.Update 2011:clinical and genetic issues in familial dilated cardiomyopathy[J].Am Coll Cardiol,2011,57(16):1641-1649.
[7]Guo W,Bharmal SJ,Esbona K,et al.Titin diversity-alternative splicing gone wild[J].Biomed Biotechnol,2010,2010(1):753675.
[8]Herman DS,Lam L,Taylor MRG,et al.Truncations of titin causing dilated cardiomyopathy[J].N Engl J Med,2012,366(7):619-628.
[9]Ceyhanbirsoy O,Agrawal PB,Hidalgo C,et al.Recessive truncating titin gene,TTN,mutations presenting as centronuclear myopathy[J].Neurology,2013,81(14):1205-1214.
Mutation screening for the causative gene in a dilated cardiomyopathy pedigree of Hubei province.
LV Yong-nan1,XU Hong-xin1,WANG Jing-wei2,ZHANG Zong-wei1.Department of Cardiology1,Department of Clinical Laboratory2,Renmin Hospital of Wuhan University,Wuhan 430060,Hubei,CHINA
ObjectiveTo screen the candidate pathogenic gene among family members of a dilated cardiomyopathy(DCM),and find the relationship between the genotype and the phenotype.MethodsThe proband and family members came from Daye City,Hubei Province.The proband was diagnosed with dilated cardiomyopathy at Renmin Hospital of Wuhan University in April 2017,and some of his family member were dead then.The inheritance atlas was drawn,and analysis of genetic characteristics and clinical phenotype was performed.The candidated gene exon of the proband was sequenced by high-throughput sequencing,and ultimately the target area of the exon and mutations of candidate genes were screened.Then bidirectional sequencing of Sanger was used to sequence other family members which were matching with gender and age to testify whether there are the above mutations.ResultsIn this family,the proband and his sister carry one missense mutations,with TTN c.100126A>G(p.Thr33376Ala).The heart function of proband was declined and accompanied by malignant arrhythmia.But his sister has no obvious clinical symptoms.ConclusionThe patient of this family carries the genetic mutation of TTN.TTN truncating mutations are a common cause of dilated cardiomyopathy,which maybe improve our understanding of the pathophysiology of dilated cardiomyopathy.
Dilated cardiomyopathy(DCM);Gene mutation;TTN
R542.2
A
1003—6350(2017)22—3627—03
国家自然科学基金(编号:81170307)
徐红新。E-mail:303876340@qq.com
2017-05-29)
