烃分子在H-FAU分子筛上吸附模拟研究
2017-12-06凤孟龙田辉平
凤孟龙,龙 军,周 涵,田辉平,赵 毅
(中国石化石油化工科学研究院,北京 100083)
烃分子在H-FAU分子筛上吸附模拟研究
凤孟龙,龙 军,周 涵,田辉平,赵 毅
(中国石化石油化工科学研究院,北京 100083)
利用量子力学与分子力学结合的方法研究C14,C22,C30的直链2-烯烃、芳烃、直链烷烃、环烷烃在H-FAU分子筛上的吸附特性,烃分子吸附能随碳数的增加而增大。烃分子吸附在H-FAU分子筛上,烯烃和芳烃双键碳原子因具有π电子与分子筛B酸中心间形成π-H键,烷烃、环烷烃与B酸中心间有电子诱导作用,烃分子与分子筛骨架间有范德华作用。烃分子碳数的增加会增大分子与分子筛骨架间的范德华作用,不影响烯烃、芳烃与B酸中心间π-H键作用或烷烃、环烷烃与B酸中心间电子诱导作用。不同类型烃分子与分子筛B酸中心间相互作用由大到小的顺序为直链2-烯烃>芳烃>直链烷烃>环烷烃。
吸附能 π-H键 电子诱导作用 范德华作用
催化裂化反应中难裂化的稠环芳烃对易裂化组分的竞争吸附效应会降低催化剂活性中心利用率,导致原料油转化率和轻质油收率降低[1]。增加剂油比,可以增加活性中心数量消除竞争吸附效应,但受到装置热平衡的限制,剂油比不能过高。过高剂油比还会造成过度裂化导致干气、焦炭产率上升,汽油、柴油等轻质油收率降低[2]。因此应对烃分子在分子筛上吸附进行研究,寻找削弱竞争吸附效应的方法,使易裂化烃分子能更容易与活性中心接触,进行裂化反应,提高催化裂化原料油的转化率和轻质油收率。现阶段关于烃分子在分子筛上的吸附研究方法主要有实验法(微量热法[3]、重量法[4]、色谱法[5]、程序升温脱附法[6]、红外光谱法[7]和核磁共振法[8]等)和分子模拟法(分子力学法[9]、分子动力学法[10]、蒙特卡罗法[11]、量子力学法和分子力学结合法[12])。利用分子模拟法,在获得宏观吸附数据的同时,还能获得吸附构型、吸附时发生的电子转移、化学键变化等数据,在研究烃分子在分子筛吸附有更广泛的应用。本课题通过量子力学和分子力学结合法研究C14,C22,C30的直链2-烯烃、芳烃、直链烷烃、环烷烃在H-FAU分子筛上的吸附特性。
1 模型和模拟方法
1.1 模型化合物的选择
以C14,C22,C30的直链烷烃、直链2-烯烃、环烷烃、芳烃为模型化合物,利用 Materials Studio8.0中基于密度泛函理论的Dmol3模块,对模型化合物的立体构型、电子结构进行优化。计算精度选择Fine,选用广义梯度近似GGA的PBE泛函,机组选用DNP,自洽场(SCF)迭代收敛的阀值设为:能量收敛精度1.0×10-5Ha,受力收敛精度0.000 2 Ha/nm,位移收敛精度0.000 5 nm。
1.2 H-FAU型分子筛模型的建立
建立636T(2022个原子)的具有FAU分子筛完整正弦孔道的模型,不饱和的Si原子用H原子饱和,Si—H键长固定为0.147 nm。将模型中一个Si原子替换成Al原子,并在与之相连的位于六方棱柱的四元环上O1位置[13-14]的O原子增加一个H原子作为电荷补偿,形成B酸中心,见图1。选取模型中40T(107个原子,球棍模型显示区域),规定为量子力学区域,模型中其余原子为分子力学区域(线性模型显示区域),利用Qmera模块对分子筛模型进行优化,计算精度选择Fine。

图1 H-FAU分子筛模型○—H; ●—O; ●—Si; ●—Al
1.3 吸附能计算
利用Qmera模块对吸附构型进行结构优化,优化后得到吸附构型能量Eadsorption,精度选择Fine。利用Qmera模块计算吸附前分子筛的能量Ezeolits和烃分子的能量Emoleculars,根据式(1)计算吸附能Eads:
Eads=Eadsorption-Ezeolits-Emoleculars
(1)
式中:Eads为吸附能;Eadsorption为吸附后吸附构型能量;Ezeolits为吸附前分子筛能量;Emoleculars为吸附前烃分子能量。
1.4 π-H键作用能、电子诱导能、范德华作用能的计算

(2)

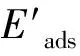
根据式(3)计算Eπ -H(或Eind):
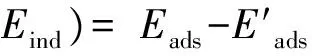
(3)
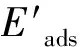
2 结果与讨论
2.1 烯烃在H-FAU分子筛上的吸附
直链2-烯烃分子吸附在H-FAU分子筛时,烯烃双键与B酸中心形成π-H键,烯烃与分子筛骨架间存在长程的范德华作用,因此吸附能(Eads)可以分解为π-H键作用能(Eπ -H)和范德华作用能(Evdw)。计算不同碳数的烯烃在H-FAU 分子筛的Eads,Eπ -H,Evdw,结果见表1。由表1可知:随着碳数的增加烯烃分子的总吸附能增大;比较Eπ -H和Evdw可以看出,Eπ-H随着碳数的增加基本不变,而Evdw随着碳数增加显著增大。这表明烯烃分子碳数的增加并不会增强烯烃分子双键与B酸中心之间的π-H键强度而是增强了烯烃分子与分子筛骨架之间的范德华相互作用。比较烯烃分子吸附前后H-FAU分子筛上H1原子的电荷和O1—H1键长变化,见图2和表1。从表1可以看出,2-烯烃分子吸附在H-FAU上形成π-H键时,分子筛上的H1原子得到电子,O1—H1键伸长。不同碳数2-烯烃分子吸附时分子筛H1原子得电子数和O1—H1键长变化量也基本相等。

表1 直链2-烯烃在H-FAU上的吸附
2.2 芳烃在H-FAU分子筛上的吸附
芳烃分子吸附在H-FAU分子筛上时,芳烃π电子与B酸中心同样形成π-H键,同时芳烃与分子筛骨架间存在长程的范德华作用。计算不同碳数的烷基苯在H-FAU 分子筛的Eads、Eπ -H和Evdw,结果见表2。由表2可以看出:随碳数的增加烷基苯的Eads增大;比较Eπ -H和Evdw可以看出,随着碳数的增加Eπ -H基本保持不变,而Evdw显著增大。这表明烷基苯分子中侧链碳数的增加并不会增强烷基苯与活性中心之间的π -H键强度而是增强了烷基苯与分子筛骨架之间的范德华相互作用。分析烷基苯分子吸附前后H-FAU分子筛H1原子的电荷和O1—H1键长变化,结果见图3、表2。烷基苯在H-FAU上吸附形成π -H键时,分子筛上的H1原子得到电子,O1—H1键伸长。不同碳数烷基苯分子吸附时分子筛H1原子得电子数和O1—H1键长变化量也基本相等。

表2 烷基苯在H-FAU上的吸附
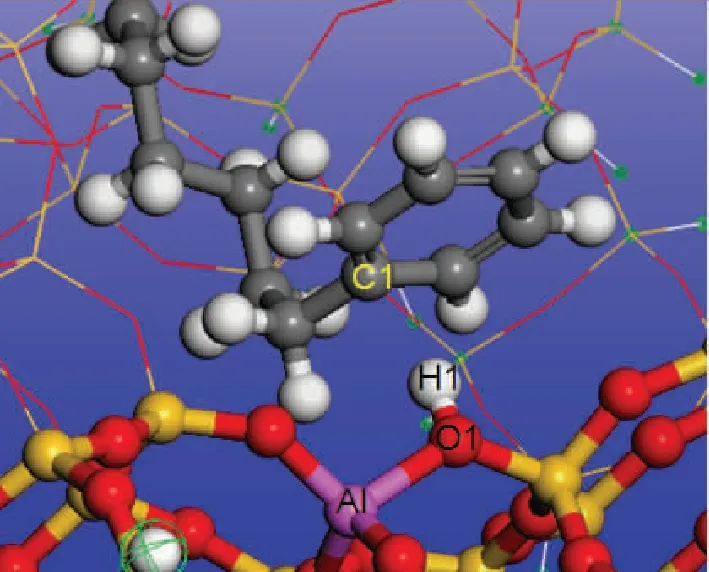
图3 烷基苯在H-FAU分子筛上的吸附示意
计算不同碳数的烷基萘和烷基蒽在H-FAU分子筛上的Eads,Eπ -H,Evdw,结果见表3,吸附示意见图4、图5。由表3可知:随着碳数的增加,烷基萘和烷基蒽在H-FAU分子筛上Eads增大;比较Eπ -H和Evdw可以看出,烷基萘和烷基蒽与分子筛B酸中心间的Eπ -H随着碳数的增加基本不变,而Evdw随着碳数增加显著增大。这表明侧链碳数的增加并不会增强烷基萘和烷基蒽与B酸中心之间的π-H键作用,而是增强与分子筛骨架之间的范德华相互作用。从表3、图4、图5可以看出,不同碳数烷基萘和烷基蒽吸附时,分子筛上H1原子得电子数、O1—H1键长变化量基本相等。

表3 烷基萘、烷基蒽在H-FAU上的吸附
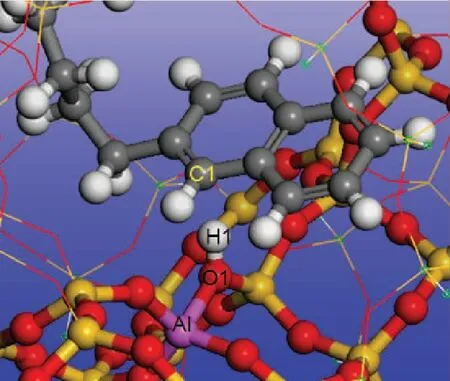
图4 烷基萘在H-FAU分子筛上的吸附示意

图5 烷基蒽在H-FAU分子筛上的吸附示意
比较烷基苯、烷基萘和烷基蒽在H-FAU吸附时的Eπ -H可以看出:随着芳烃环数的增加,π-H键作用降低;进一步比较芳烃吸附时分子筛上H1原子的得电子数和O1—H1键长变化量可以看出,芳环数量的增加,芳烃在分子筛B酸中心上吸附时H1原子的得电子数和O1—H1键长变化量均降低。
2.3 直链烷烃在H-FAU分子筛上的吸附
直链烷烃分子吸附在H-FAU分子筛上时,分子筛B酸中心通过诱导作用使分布在直链烷烃C—H键间的电子云发生极化从而得到部分电子,形成电子诱导作用(Eind),直链烷烃与分子筛骨架间也存在长程的范德华作用(Evdw)。吸附能(Eads)可以分解为诱导作用能(Eind)和范德华作用能(Evdw)。计算不同碳数的直链烷烃在H-FAU 分子筛的Eads,Eind,Evdw,结果见表4。由表4可知:随着碳数增加,烃分子的总吸附能增大;比较Eind和Evdw可以看出,Eind随碳数增加变化不大,而Evdw随碳数增加显著增大。这表明碳数的增加并不会增强直链烷烃分子与B酸中心之间的电子诱导作用而是增强了直链烷烃分子与分子筛骨架之间的范德华相互作用。分析直链烷烃分子吸附前后H-FAU分子筛上H1原子的电荷和O1—H1键长变化,如图6、表4所示,烃分子吸附在H-FAU上时,分子筛上的H1原子得到电子,O1—H1键伸长。不同碳数直链烷烃吸附时,分子筛H1原子得电子数和O1—H1键长变化量也基本相等。

表4 直连烷烃在H-FAU分子筛上的吸附
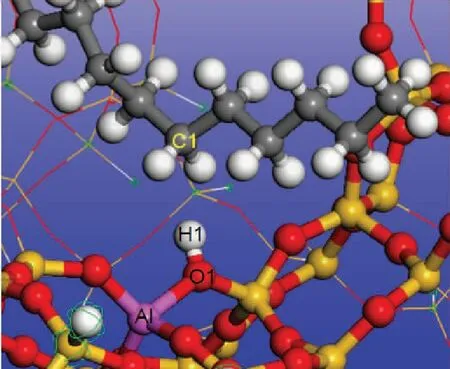
图6 直链烷烃在H-FAU分子筛上的吸附示意
2.4 环烷烃在H-FAU分子筛上的吸附
烷基环己烷吸附在H-FAU分子筛上时,与直链烷烃类似,吸附能(Eads)可以分解为诱导作用能(Eind)和范德华作用能(Evdw)。计算不同碳数的烷基环己烷吸附在H-FAU 分子筛的Eads,Eind,Evdw,结果见表5。由表5可知:随着碳数的增加Eads增大;Eind随碳数增加基本保持不变,而范德华作用Evdw随碳数增加显著增大。这表明烷基环己烷侧链碳数的增加并不会增强烷基环己烷与B酸中心之间的电子诱导作用而是增强了烷基环己烷与分子筛骨架之间的范德华作用。分析烷基环己烷吸附前后H-FAU分子筛上H1原子的电荷变化和O1—H1键长变化,如表5、图7所示,烷基环己烷在H-FAU上吸附时,分子筛上的H1原子得到电子,O1—H1键伸长。不同碳数烷基环己烷吸附时,分子筛H1原子得电子数和O1—H1键长变化量也基本一致。

表5 烷基环己烷在H-FAU分子筛上的吸附

图7 烷基环己烷在H-FAU分子筛上的吸附示意
计算不同碳数的烷基萘烷、烷基全氢蒽在H-FAU分子筛上的Eads,Eind,Evdw,结果见表6。由表6可知:随着碳数的增加,烷基萘烷和烷基全氢蒽在H-FAU分子筛上Eads增大;比较Eind和Evdw可以看出,烷基萘烷和烷基全氢蒽与分子筛活性中心间的Eind随着碳数的增加基本保持不变,而Evdw随着碳数增加显著增大;这表明侧链碳数的增加并不会增强烷基萘烷和烷基全氢蒽与B酸中心之间的电子诱导作用而是增强与分子筛骨架之间的范德华作用。烷基萘烷、烷基全氢蒽在H-FAU分子筛上的吸附示意见图8、图9。从图8、图9、表6可以看出,不同碳数烷基萘烷和烷基全氢蒽吸附时,分子筛上H1原子得电子数、O1—H1键长变化量基本相等。

表6 烷基萘烷、烷基全氢蒽在H-FAU上的吸附

图8 烷基萘烷在H-FAU分子筛上的吸附示意

图9 烷基全氢蒽在H-FAU分子筛上的吸附示意
比较烷基环己烷与烷基萘烷和烷基全氢蒽与分子筛间的Eind,烷基萘烷和烷基全氢蒽的Eind大于烷基环己烷。比较环烷烃吸附时分子筛上H1原子的得电子数和O1—H1键长变化量可以看出,烷基萘烷和烷基全氢蒽吸附时分子筛H1原子得到电子数和O1—H1键长变化量均大于烷基环己烷。
2.5不同类型烃分子吸附比较
比较直连2-烯烃、芳烃、直连烷烃和环烷烃在H-FAU上的吸附可以看出,不同类型烃分子吸附能均随着碳数的增加而增大,但碳数的变化对烃分子与分子筛B酸中心之间的相互作用能影响较小。不同类型烃分子与分子筛B酸中心间相互作用强度存在较大差异,烯烃、芳烃由于其π电子与B酸中心形成π-H键作用,与活性中心间相互作用较强。芳烃分子与B酸中心间相互作用弱于烯烃。直连烷烃、环烷烃与B酸中心间具有电子诱导作用,弱于烯烃、芳烃与活性中心形成的π-H键作用。直链烷烃与B酸中心间的电子诱导作用强于环烷烃。环烷烃中,烷基环己烷与B酸中心间电子诱导作用弱于烷基萘烷、烷基全氢蒽。不同类型烃分子与B酸中心相互作用强度顺序为:烯烃>芳烃>直链烷烃>环烷烃。
3 结 论
(1)烃分子吸附在H-FAU分子筛上,烯烃、芳烃、直连烷烃、环烷烃分子吸附能均随着分子碳数的增加逐渐增大。
(2)烯烃、芳烃、直连烷烃、环烷烃分子吸附在H-FAU上,分子碳数的增加只会增加烃分子与分子筛骨架间的范德华作用,对B酸中心与烃分子间的π-H键作用或电子诱导作用影响较小。
(3)烃分子与分子筛B酸中心间的相互作用强弱顺序为:烯烃>芳烃>直链烷烃>环烷烃。
[1] 陈小博,辛利,李楠,等.焦化蜡油中芳香分的催化裂化特性及其对饱和分裂化性能的阻滞作用[J].中国石油大学学报,2014,38(5):190-195
[2] 刘银东,李泽坤,王刚,等.竞争吸附对催化裂化反应过程的影响[J].化工学报,2008,59(11):2794-2799
[3] Coker E N,Jia Chunjuan,Karge H G.Adsorption of benzene and benzene derivatives onto zeolite H-Y studied by microcalorimetry[J].Langmuir ,2000,16(3):1205-1210
[4] Makowski W,Majda D.Equilibrated thermodesorption studies of adsorption of n-hexane and n-heptane on zeolitesY,ZSM-5 and ZSM-11[J].Applied Surface Science,2005,252(3):707-715
[5] Denayer J F,Souverijns W,Jacobs P A,et al.High-temperature low-pressure adsorption of branched C5—C8alkanes on zeolite beta,ZSM-5,ZSM-22,zeolite Y,and mordenite[J].Phys Chem B ,1998,102(23):4588-4597
[6] Makowski W ,Ogorzalek L.Determination of the adsorption heat ofn-hexane andn-heptane on zeolites beta,L,5A,13X,Y and ZSM-5 by means of quasi-equilibrated temperature-programmed desorption and adsorption(QE-TPDA)[J].Thermochimica Acta ,2007,465(12):30-39
[7] Benaliouche F,Boucheffa Y,Thibault-Starzyk F.In situ FTIR studies of propene adsorption over Ag- and Cu-exchanged Y zeolites[J].Microporous and Mesoporous Materials,2012,147(1):10-16
[8] Zhu Jianfeng,Mosey N,Woo T.Study of the adsorption of toluene in zeolite LiNa-Y by solid-state NMR spectroscopy[J].Phys Chem C,2007,111(36):13427-13436
[9] Nascimento M A C.Computer simulations of the adsorption process of light alkanes in high-silica zeolites[J].Journal of Molecular Structure(Theochem),1999,464(123):239-247
[10] Krishna R,Baten J M.Diffusion of hydrocarbon mixtures in MFI zeolite:Influence of intersection blocking[J].Chemical Engineering Journal,2008,140(1):614-620
[11] Khettar A ,Jalili S E ,Dunne L J.Monte-carlo simulation and mean-field theoryinterpretation of adsorption preference reversal in isotherms of alkane binary mixtures in zeolites at elevated pressures[J].Chemical Physics Letters,2002,362(56):414-418
[12] Raksakoon C,Limtrakul J.Adsorption of aromatic hydrocarbon onto H-ZSM-5 zeolite investigated by Oniom study[J].Journal of Molecular Structure(Theochem),2003,631(123):147-156
[13] Liengme B V,Hall W K.Studies of hydrogen held by solids.Part 11—Interaction of simple olefins and pyridine with decationated zeolites[J].Trans Farady Soc,1966,62:3229-3243
[14] Olson D H ,Dempsey E.The crystal structure of the zeolite hydrogen faujasite[J].Journal of Catalysis ,1969 ,13(2):221-231
SIMULATIONSTUDYOFHYDROCARBONADSORPTIONONH-FAUZEOLITEWTBZ
Feng Menglong, Long Jun, Zhou Han, Tian Huiping, Zhao Yi
(SINOPECResearchInstituteofPetroleumProcessing,Beijing100083)
Molecular simulation methods were used to study the adsorption of different types of hydrocarbons (2-olefins,aromatics,alkanes and cycloalkanes with carbon chain of C14,C22,C30) on H-FAU zeolite.The results show that the adsorption energy increases as the chain length extends from 14 to 30.The π-H bond is formed between the Bronsted acid sites on the zeolite and the π-electronics of olefins and aromatics,while Van der Waals interaction exists between zeolite framework and molecules through the electronic induction between Bronsted acid sites and σ-electronics of alkanes and cycloalkanes.The increase of carbon number in molecular chain has little effect on π-H interaction or electronic induction interaction,but increases the Van der Waals interactions.The intensity of interactions between molecules and acid sites decreases in the following order:olefins>aromatics>alkanes>cycloalkanes.
adsorption energy; π-H bond;electronic induction; Van der Waals interaction
2017-04-21;修改稿收到日期2017-08-09。
凤孟龙,博士研究生,主要研究VGO分子在FAU分子筛上的扩散和吸附过程。
周涵,E-mail:zhouhan.ripp@sinopec.com。
中国石油化工股份有限公司合同项目(No.YK515026)。
