原发性胆汁性胆管炎的遗传学研究现状
2017-11-22李奕康唐茹琦
李奕康, 马 雄, 唐茹琦
(上海交通大学医学院附属仁济医院 消化内科, 上海市消化病研究所, 上海 200001)
原发性胆汁性胆管炎的遗传学研究现状
李奕康, 马 雄, 唐茹琦
(上海交通大学医学院附属仁济医院 消化内科, 上海市消化病研究所, 上海 200001)
原发性胆汁性胆管炎(PBC)是一种具有明显遗传易感性的自身免疫性肝病。近年来开展的全基因组关联分析和基因芯片等遗传学研究,在揭示PBC的发病机制中发挥着重要的作用。PBC的易感基因主要分为人类白细胞抗原基因和非人类白细胞抗原基因两大类,其易感位点大多与免疫调节相关,表明免疫调节通路紊乱可能在PBC的发病中起关键作用。此外,这些候选基因的加权基因风险评分在一定程度上可能预测PBC的发病风险。如今,约有1/3的PBC患者对现有的PBC标准治疗药物熊去氧胆酸反应不佳。因此,易感基因对应的靶向药物有望成为有效的替代疗法。
胆管炎, 胆汁性; 胆汁淤积; 疾病遗传易感性
原发性胆汁性胆管炎(PBC),原名为“原发性胆汁性肝硬化”,是一种具有明显遗传易感性的,以肝内小叶间胆管非化脓性炎症损伤及血清中高滴度抗线粒体抗体为特征的慢性进行性胆汁淤积性肝病[1]。在40岁以上的妇女中,PBC的发病率约为1/1000,并呈逐年升高的趋势[2]。我国PBC的发病率和病死率均明显高于世界其他地区[3]。因此,明确其遗传学发病机制,制定在根源上防治PBC的策略至关重要。目前,在揭示PBC的发病机制,筛查PBC高危人群的遗传学研究中,全基因组关联分析(genome-wide association study,GWAS)和基因芯片(immunochip studies,ICHIP)等研究发挥着重要的桥梁作用[4]。其中,GWAS能够以基因组中的单核苷酸多态性(single nucleotide polymorphism, SNP)为分子遗传标记,在全基因组水平上进行对照分析或相关性分析,从而发现与PBC相关的遗传变异;而ICHIP则涵盖186个已知的自身免疫性疾病的易感区域上的196 524个遗传多态性,能够深入剖析这些区域内常见和罕见的遗传变异与PBC的关联[5]。现就PBC的遗传学研究进展进行综述。
1 遗传学研究
家系调查[6]表明,PBC患者同胞的患病风险比正常人高9.5倍,其一级亲属的患病率比正常人高5.8~9.7倍。此外,在双胞胎PBC患者中,同卵双胞胎的同病率高达63%,而异卵双胞胎的同病率为0,提示PBC具有明显的遗传倾向。GWAS等遗传学研究进一步证实了遗传因素与PBC的发病密切相关。现已发现的PBC易感基因主要有人类白细胞抗原(human leukocyte antigen,HLA)基因和非HLA(non-HLA)基因2大类。
1.1 HLA基因多态性 HLA是现已证实的与PBC发病关系最密切的基因[7-9]。HLA复合体位于6p21,可以表达自身抗原,参与抗原递呈,诱导免疫耐受和参与免疫调节。根据HLA的结构及其特点,HLA复合体分为Ⅰ、Ⅱ、Ⅲ类基因区。而现有研究表明,参与PBC发病的主要是HLA-Ⅱ类基因。其中,相关性最强的是HLA-DRB1、HLA-DQA1和HLA-DQB1基因[8-10]。在欧洲和北美高加索人群中,DRB1*0801与PBC相关最为常见[11-12];在日本人群中,DRB1*0803提示与PBC相关联[13];在我国人群中,PBC患者出现DRB1*07、DRB1*08,DRB1*0801的频率较健康对照组显著升高[14-15],提示PBC的遗传易感性存在种族差异。荟萃分析[16]显示,HLA-DR*07、08基因是PBC的危险因素,而DR*11、*12、*13和*15则可能是保护因素。其中,HLA-DRB1等位基因的多态性所编码的不同DRB1氨基酸残基,可能通过影响对抗原的结合亲和力,进而影响PBC的遗传易感性[17];保护性的遗传位点DRB1*11和DRB*13在对抗HCV、HIV和人类乳头瘤病毒中也具有重要作用,提示PBC的发病机制还可能与病原体感染有关[18]。尽管HLA基因与PBC发病关系密切,但有80%~90%的PBC患者并未携带这些常见的HLA基因变异,提示在non-HLA基因中可能也存在与PBC发生发展相关的基因位点。
1.2 non-HLA基因多态性 目前GWAS、ICHIP等9项大型研究[19-27]共发现了30多个non-HLA基因位点与PBC相关(表1)。这些GWAS研究在基因组水平发现了许多与PBC相关的易感位点和生物学通路,加深了对PBC遗传结构的认识,进一步揭示了遗传因素在PBC发生发展中的重要作用。此外,non-HLA基因在PBC的发病中也具有极其重要的作用。这些基因大多已证实与其他自身免疫性疾病相关,并有可能参与包括PBC在内的自身免疫性疾病的免疫调节紊乱[8]。其中,IL-12A及IL-12RB2是在GWAS研究中被证实与PBC关联最紧密的non-HLA基因位点[28],提示IL-12免疫调节信号通路相关基因可能在PBC的发病中发挥着极其重要的作用。IL-12细胞因子家族是一组共享蛋白质链的异二聚体分子,包括IL-12、IL-23、IL-27、IL-35,其主要涉及IL-12/Th1通路和IL-23/Th17通路。其中,IL-12通过刺激Th1细胞的免疫应答[28],主要参与PBC的发生;而IL-23/Th17细胞信号通路则是PBC进展的重要原因[29]。在被证实与PBC相关的基因中,至少有10个基因与IL-12相关免疫调节通路直接或间接相关——干扰素调节因子(interferon regulatory factors,IRF)5、细胞因子信号转导抑制蛋白(suppressor of cytokine signaling,SOCS)1、核因子-κB(NF-κB)1和肿瘤坏死因子超家族(tumor necrosis factor superfamily,TNFSF)1A均参与IL-12的产生[30],IRF8和TNFSF15通过其编码产物调节IL-12及INFγ的产生[21,30],IL-12A和IL-12RB2分别是编码IL-12及其受体的基因,酪氨酸激酶(tyrosine kinase,TYK)2和信号转导及转录激活因子(signal transducers and activators of transcription,STAT)4则被证实可影响IL-12信号通路的下游[31]。然而,由于IL-12家族的复杂性以及对其涉及通路功能的认识不充分,IL-12相关通路在PBC发病发展中的确切机制尚不明确。
此外,在与PBC发病相关的non-HLA基因中,TNFRSF1A、CD80、RPS6KA4、MAP3K7IP1和NF-κB这5个候选基因可能与NF-κB信号通路有关。其中,TNFRSF1A是编码肿瘤坏死因子受体(tumor necrosis factor receptor,TNFR)1的基因,已被证实与多种自身免疫性疾病有关[32],可通过调控Kupffer细胞及肝星状细胞的激活,导致肝纤维化。在动物实验[33]中,TNFRSF1A-/-小鼠还被证实可以减轻二甲基亚硝胺导致的肝纤维化。CD80是位于抗原递呈细胞表面的共刺激分子,与T淋巴细胞表面的CD28相互作用,可激活幼稚CD4+Th0淋巴细胞,并使之在IL-12和IL-12R的作用下分化成Th1淋巴细胞,参与免疫应答[34]。RPS6KA4可以抑制Toll样受体依懒性细胞因子的表达[24],影响调节性T淋巴细胞的功能,破坏免疫耐受,诱导自身免疫反应。MAP3K7IP1可以编码丝氨酸/苏氨酸蛋白激酶,是参与细胞转录和凋亡相关信号转导通路的重要组成成分[35]。值得注意的是,TNFRSF1A、CD80、RPS6KA4和MAP3K7IP1还是参与NF-κB活化的基因[24];而NF-κB本身是参与多种免疫调节基因表达的转录因子,并在类风湿性关节炎和多发性硬化等自身免疫疾病中高度活化。同时,NF-κB p50-/-小鼠表现出进展性的肝脏炎症和纤维化,NF-κB还参与调节活化肝星状细胞存活和凋亡的平衡[36],均提示NF-κB信号通路可能在PBC的发病中具有重要作用。此外,Qiu等[19]在我国汉族人群中证实了IL-21、IL-21R、CD28/CTLA4/ICOS、ARID3A及IL-16等基因与PBC发病相关,并通过组织学研究发现PBC患者肝脏内IL-21和IL-21R表达显著上升,IL-21+细胞水平及IL-21R+细胞水平均与肝炎症程度和纤维化程度呈正相关,表明IL-21免疫调节通路、CD4 T淋巴细胞的激活和T淋巴细胞共刺激也可能是PBC发展的重要因素。总之,多种non-HLA基因及其对应的信号通路可能通过免疫调节参与PBC的发病,但其确切功能仍有待进一步研究。
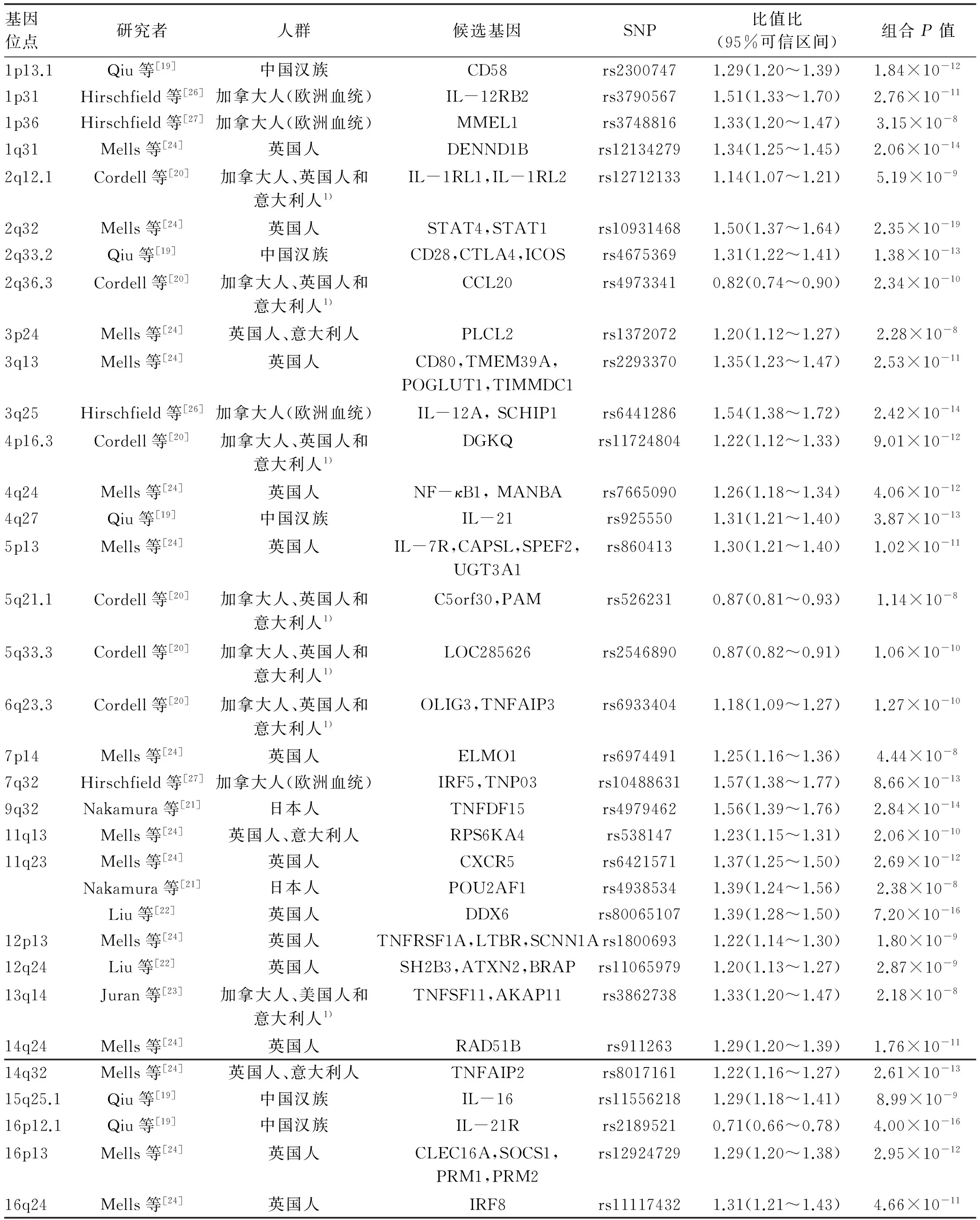
表1 与PBC显著相关的non-HLA基因位点(P<5×10-8)

基因位点研究者人群候选基因SNP比值比(95%可信区间)组合P值17q12 Liu等[22] 英国人 ORMDL3,ZPBP2,GSDMB,IKZF3rs8067378 1.26(1.19~1.34) 6.05×10-14 17q21Liu等[22]英国人MAPT,CRHR1rs175648291.25(1.16~1.35)2.15×10-919p12Liu等[22]英国人TYK2rs345364431.91(1.59~2.28)1.23×10-1219p13.3Qiu等[19]中国汉族ARID3Ars104159760.77(0.72~0.84)3.61×10-1219q13Liu等[25]意大利人SPIBrs37455161.462)7.97×10-1122q13Mells等[24]英国人MAP3K7IP1rs9684511.27(1.18~1.38)1.08×10-9Liu等[22]英国人SYNGR1,PDGFB,RPL3rs22674071.29(1.21~1.38)1.29×10-13
注:1)研究人群均为欧洲血统;2)95%可信区间无法获取
1.3 其他遗传学因素 研究[37]表明,PBC患者与健康人群相比X染色体单体率上升,Y染色体丢失率增加,提示PBC的发病还可能与性染色体缺失有关。此外,Selmi等[38]发现同卵双胞胎PBC患者和正常同卵双胞胎相比,有60个甲基化差异区域,其中51个位于X染色体,可能与PBC好发于女性有关。同时,有研究[39]发现PBC患者自身免疫性T淋巴细胞的CD40L、LIGHT、IL-17和IFNγ等基因启动子区域组蛋白H4乙酰化,且血清中存在81种差异表达的miRNA[40]。因此,DNA甲基化、组蛋白修饰及非编码RNA干扰等表观遗传修饰也可能参与PBC的发病。
1.4 遗传学研究的局限性 尽管遗传学研究的开展发现了越来越多与PBC相关的基因,但这些基因仅能够解释不足20%PBC患者的遗传性[22,41]。其原因可能是相关研究的样本数较少,风险基因座内等位基因的异质性较大[17],GWAS等技术无法检测到可能具有强烈效应的低频、罕见变异[42]及非SNP结构改变[31](如基因拷贝数的改变、表观遗传修饰及基因与环境因素的相互作用)。此外,GWAS研究的芯片中检测到的SNP尚不能覆盖基因组所有的SNP,很多研究是根据SNP之间的连锁不平衡挑选tag SNP进行检测。因此,GWAS研究所发现相关联的SNP在多数情况下并不是疾病真正的易感位点,而是与致病位点处于高度连锁不平衡。因此,仅根据GWAS研究并不能发现PBC的所有易感基因和真正的致病位点。
如今,随着外显子和全基因组的新一代基因测序技术的发展以及成本的不断下降,通过大规模测序来鉴定PBC的遗传位点成为可能。然而,基因组测序并不能直接反映PBC发生发展过程中RNA水平和蛋白质水平的变化,所以在确定易感基因之后仍需进行功能研究。因此,基于转录组学和蛋白质组学的大样本研究具有重要的价值[43]。综上所述,虽然GWAS研究通过在基因组水平确定了许多PBC的易感位点和相关的生物学通路,但仍需要进一步精细定位易感基因和对其进行生物学功能研究。为揭示PBC遗传学发病机制,新一代基因测序技术以及将遗传学、表观遗传学、转录组学、蛋白质组学、代谢组学和临床中间表型整合到GWAS研究中可能是未来研究的趋势。
2 临床应用
GWAS等遗传学研究加深了对PBC遗传结构的认识,但是如何把所得的遗传学信息应用于临床仍在探索之中。现有的尝试主要是应用易感位点进行PBC的患病风险评估以及为熊去氧胆酸(UDCA)治疗不佳的PBC患者寻找潜在的替代治疗方案。
2.1 风险评估 为尝试将PBC现有的遗传信息应用于临床,Dong等[44]利用26个PBC易感位点制作加权基因危险风险评分,在识别个体发生PBC的风险中取得较好的预测效果,受试者工作特征曲线下面积为0.82。最高风险组比最低风险组的PBC发病风险增加了9.31倍。因此,随着临床测序的普及其预测值准确度的提高,利用遗传信息有望在诊断PBC及筛查高危人群中起到重要的作用。
迄今为止,对于PBC风险评估的遗传学研究主要集中在筛查发病相关的遗传变异,鲜有对治疗应答及其并发症相关的遗传学研究,而后者对于PBC临床风险分层和预后有更重要的意义。有研究[45]对接受UDCA治疗的PBC患者开展预后评分,其预测值有一定的价值,阳性者5年和10年生存率分别为98%和88%,但该评分标准却未包括遗传因素,表明遗传学信息在PBC临床风险分层和预后评分中的作用尚未被发掘。因此,开展与PBC治疗应答相关的多中心大样本大型遗传学研究,对于PBC患者选择最佳的治疗方案与时机有重要意义。
2.2 替代治疗
如今,PBC的一线治疗药物主要是UDCA。作为一种无毒性的次级胆汁酸,UDCA具有促进内源性胆汁酸排泄,拮抗疏水性胆酸细胞毒性,保护肝细胞,抑制内源性胆固醇合成,促进其代谢,参与免疫调节和抑制炎症反应等作用。因此,UDCA可以改善大部分PBC患者的肝功能,延缓肝脏组织学进展,减少对肝移植的需求。但仍然有约1/3的PBC患者对UDCA的治疗反应不佳或者没有应答,从而导致病情进展。因此,亟需找到新的替代治疗方案。现有的补充治疗药物中,奥贝胆酸(OCA)可显著降低对UDCA应答不佳患者血清GGT、ALP、ALT及TBil水平[46],已被美国食品药品监督管理局批准为新的PBC治疗药物。OCA可联合UDCA应用于对UDCA治疗不佳者,也可单独用于不能耐受UDCA治疗者[47],但OCA并不能改善PBC患者的肝纤维化水平,还会增加皮肤瘙痒及严重不良事件的发生率[48]。而其他治疗药物如非诺贝特[49-50]、环孢霉素A[51]等疗效尚不尽人意。研究表明,PBC的易感基因可能是新的治疗靶点,并有望成为现有治疗方案的重要补充。
2.2.1 利妥昔单抗 利妥昔单抗是B淋巴细胞表面分子CD20的特异性抗体,现已应用于多种自身免疫性疾病的治疗。在已知的PBC候选基因中,POU2AF1和IKZF3可编码促进B淋巴细胞成熟的转录因子,并促进生发中心的形成;IL-7R参与前B淋巴细胞的增殖;ARID3A则是一种B淋巴细胞限制性的转录因子,可激活免疫球蛋白重链基因转录,并参与自身免疫反应[52]。因此,B淋巴细胞可能在PBC的发病中起到重要作用,为利妥昔单抗用于治疗PBC提供了重要的理论依据。现已证实,利妥昔单抗可以明显降低对UDCA治疗不佳的PBC患者体内自身抗体水平[53]。然而,在PBC小鼠模型中,利妥昔单抗却导致了更为严重的胆管炎,引发了对其治疗安全性的担忧[54]。因此,未来仍需要进一步探究利妥昔单抗对PBC的治疗效果及安全性。
2.2.2 优斯它单抗 优斯它单抗的治疗靶向位点是IL-12的亚基IL-12 p40,该亚基是与PBC发病密切相关的IL-12信号通路的重要组成部分[31]。此外,优斯它单抗已被应用于治疗克罗恩病和银屑病等自身免疫性疾病。然而,在一项多中心的Ⅱ期试验[55]中,经过优斯它单抗的治疗,PBC患者的ALP水平、血清TBil水平及增强肝纤维化评分并未得到明显改善。
2.2.3 TNFα抑制剂 TNFα可激活细胞内的多种信号通路,在肝脏内环境平衡中有着重要作用[56]。在与PBC相关的候选基因中,与TNFα信号通路相关的有DENND1B、TNFRSF1A和TNFAIP2[21,24],但TNFα抑制剂是否对UDCA治疗不佳的PBC患者具有治疗效果尚不明确。
此外,其他潜在的治疗靶点还有IL-21和IL-16。其中,IL-21主要由辅助性滤泡T淋巴细胞(follicular helper T cells,Tfh)产生,而Tfh细胞表面的特异性标志物由PBC的易感基因CXCR5编码[24]。IL-21和其受体IL-21R的结合可以激活包括STAT1和STAT3等多种下游信号分子,而这些信号分子对于T淋巴细胞、B淋巴细胞及自然杀伤细胞的增殖分化都有重要作用。同时,与PBC显著相关的IL-12也可以诱导IL-21和IL-21+INFγ+CD4 T淋巴细胞的产生,进而参与免疫调节[57]。因此,IL-21可能是PBC的重要治疗靶点。此外,IL-16是一种T淋巴细胞趋化因子,与CD4 T淋巴细胞的积累相关,其中和性IL-16抗体在动物实验中可以预防胰腺炎和自身免疫性1型糖尿病[58],但用于治疗PBC的研究仍有待开展。
3 展望
GWAS等技术的应用与发展在进一步揭示PBC的遗传学发病机制中发挥了极其重要的作用,研究发现了许多与PBC发生发展相关的non-HLA易感位点和免疫调节信号通路,有助于实现个体化风险评估和治疗。已证实的易感基因及其加权基因风险评分有助于筛查PBC高危人群,评估其发病风险,并有望为UDCA治疗不佳的PBC患者提供新的替代治疗方案。不过,这些易感基因和生物学信号通路在PBC发病或进展中的具体作用机制仍有待进一步研究。此外,已知的遗传学信息尚未能很好的应用于临床。因此,这些基因的功能研究以及对PBC患者进行危险分层和预后分析是今后PBC遗传学研究的重要方向。随着新一代基因测序技术的开展以及遗传学、表观遗传学、转录组学、蛋白质组学、代谢组学和临床中间表型与GWAS研究的整合,将会对PBC的发病机制有更深入的认识。
[1] PUROHIT T, CAPPELL MS, et al. Primary biliary cirrhosis: pathophysiology, clinical presentation and therapy[J]. World J Hepatol, 2015, 7(7): 926-941.
[2] HIRSCHFIELD GM, SIMINOVITCH KA, et al. Genetic in PBC: what do the “risk genes” teach us? [J]. Clinic Rev Allerg Immunol, 2015, 48(2-3): 176-181.
[3] WONG GL, HUI AY, WONG VW, et al. A retrospective study on clinical features and prognostic factors of biopsy-proven primary biliary cirrhosis in Chinese patients[J]. Am J Gastroenterol, 2005, 100(10): 2205-2211.
[4] FLORES A, MAYO MJ. Primary biliary cirrhosis in 2014[J]. Curr Opin Gastroenterol, 2014, 30(3): 245-252.
[5] TRYNKA G, HUNT KA, BOCKETT NA, et al. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease[J]. Nat Genet, 2011, 43(12): 1193-1201.
[6] SELMI C, MAYO M J, BACH N, et al. Primary biliary cirrhosis in monozygotic and dizygotic twins: genetics, epigenetics, and environment[J]. Gastroenterology, 2004, 127(2): 485-492.
[7] CAREY EJ, ALI AH, LINDOR KD. Primary biliary cirrhosis[J]. Lancet, 2015, 386(10003): 1565-1576.
[8] HIRSCHFIELD GM, INVERNIZZI P. Progress in the genetics of primary biliary cirrhosis[J]. Semin Liver Dis, 2011, 31(2): 147-156.
[9] HUANG YQ. Recent advances in the diagnosis and treatment of primary biliary cholangitis[J]. World J Hepatol, 2016, 8(33): 1419-1441.
[10] JOSHITA S, UMEMURA T, TANAKA E, et al. Genetic contribution to the pathogenesis of primary biliary cholangitis[J]. J Immunol Res, 2017, 2017: 3073504.
[11] DONALDSON PT, BARAGIOTTA A, HENEGHAN MA, et al. HLA class II alleles, genotypes, haplotypes, and amino acids in primary biliary cirrhosis: a large-scale study[J]. Hepatology, 2006, 44(3): 667-674.
[12] MULLARKEY ME, STEVENS AM, MCDONNELL WM, et al. Human leukocyte antigen class II alleles in Caucasian women with primary biliary cirrhosis[J]. Tissue Antigens, 2005, 65(2): 199-205.
[13] ONISHI S, SAKAMAKI T, MAEDA T, et al. DNA typing of HLA class II genes; DRB1*0803 increases the susceptibility of Japanese to primary biliary cirrhosis[J]. J Hepatol, 1994, 21(6): 1053-1060.
[14] LIU HY, DENG AM, ZHANG J, et al. Analysis of HLA alleles polymorphism in Chinese patients with primary biliary cirrhosis[J]. Chin J Hepatal, 2005, 13(6): 410-413. (in Chinese)
刘海英, 邓安梅, 张建, 等. 原发性胆汁性肝硬化患者人类白细胞抗原等位基因多态性分析[J]. 中华肝脏病杂志, 2005, 13(6): 410-413.
[15] JIANG XH, ZHONG RQ, FANG XY, et al. Relationship between alleles of HLA-DRB and HLA-DQB1 and Chinese patients with primary biliary cirrhosis[J]. Chin J Hepatol, 2004, 12(7): 436. (in Chinese)
姜小华, 仲人前, 方晓云, 等. 原发性胆汁性肝硬化与HLA-DRB1、DQB1等位基因的相关性研究[J]. 中华肝脏病杂志, 2004, 12(7): 436.
[16] LI M, ZHENG H, TIAN QB, et al. HLA-DR polymorphism and primary biliary cirrhosis: evidence from a meta-analysis[J]. Arch Med Res, 2014, 45(3): 270-279.
[17] UMEMURA T, JOSHITA S, ICHIJO T, et al. Human leukocyte antigen class II molecules confer both susceptibility and progression in Japanese patients with primary biliary cirrhosis[J]. Hepatology, 2012, 55(2): 506-511.
[18] KUMAGI T, ABE M, IKEDA Y, et al. Infection as a risk factor in the pathogenesis of primary biliary cirrhosis: pros and cons[J]. Dis Markers, 2010, 29(6): 313-321.
[19] QIU F, TANG R, ZUO X, et al. A genome-wide association study identifies six novel risk loci for primary biliary cholangitis[J]. Nat Commun, 2017, 8: 14828.
[20] CORDELL HJ, HAN Y, MELLS GF, et al. International genome-wide meta-analysis identifies new primary biliary cirrhosis risk loci and targetable pathogenic pathways[J]. Nat Commun, 2015, 6: 8019.
[21] NAKAMURA M, NISHIDA N, KAWASHIMA M, et al. Genome-wide association study identifies TNFSF15 and POU2AF1 as susceptibility loci for primary biliary cirrhosis in the Japanese population[J]. Am J Hum Genet, 2012, 91(4): 721-728.
[22] LIU JZ, ALMARRI MA, GAFFNEY DJ, et al. Dense fine-mapping study identifies new susceptibility loci for primary biliary cirrhosis[J]. Nat Genet, 2012, 44(10): 1137-1141.
[23] JURAN BD, HIRSCHFIELD GM, INVERNIZZI P, et al. Immunochip analyses identify a novel risk locus for primary biliary cirrhosis at 13q14, multiple independent associations at four established risk loci and epistasis between 1p31 and 7q32 risk variants[J]. Hum Mol Genet, 2012, 21(23): 5209-5221.
[24] MELLS GF, FLOYD JA, MORLEY KI, et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis[J]. Nat Genet, 2011, 43(4): 329-332.
[25] LIU X, INVERNIZZI P, LU Y, et al. Genome-wide meta-analyses identify three loci associated with primary biliary cirrhosis[J]. Nat Genet, 2010, 42(8): 658-660.
[26] HIRSCHFIELD GM, LIU X, HAN Y, et al. Variants at IRF5-TNPO3, 17q12-21 and MMEL1 are associated with primary biliary cirrhosis[J]. Nat Genet, 2010, 42(8): 655-657.
[27] HIRSCHFIELD GM, LIU X, XU C, et al. Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants[J]. N Engl J Med, 2009, 360(24): 2544-2555.
[28] van WANROOIJ RL, ZWIERS A, KRAAL G, et al. Genetic variations in interleukin-12 related genes in immune-mediated diseases[J]. J Autoimmun, 2012, 39(4): 359-368.
[29] YANG CY, MA X, TSUNEYAMA K, et al. IL-12/Th1 and IL-23/Th17 biliary microenvironment in primary biliary cirrhosis: implications for therapy[J]. Hepatology, 2014, 59(5): 1944-1953.
[30] HIRSCHFIELD GM, CHAPMAN RW, KARLSEN TH, et al. The genetics of complex cholestatic disorders[J]. Gastroenterology, 2013, 144(7): 1357-1374.
[31] GULAMHUSEIN AF, JURAN BD, LAZARIDIS KN. Genome-wide association studies in primary biliary cirrhosis[J]. Semin Liver Dis, 2015, 35(4): 392-401.
[33] KITAMURA K, NAKAMOTO Y, AKIYAMA M, et al. Pathogenic roles of tumor necrosis factor receptor p55-mediated signals in dimethylnitrosamine-induced murine liver fibrosis[J]. Lab Invest, 2002, 82(5): 571-583.
[34] CHIKUMA S. CTLA-4, an essential immune-checkpoint for T-cell activation[J]. Curr Top Microbiol Immunol, 2017. [Epub ahead of print]
[35] WOLF A, BEUERLEIN K, ECKART C, et al. Identification and functional characterization of novel phosphorylation sites in TAK1-binding protein (TAB) 1[J]. PLoS One, 2011, 6(12): e29256.
[36] ELSHARKAWY AM, OAKLEY F, LIN F, et al. The NF-kappaB p50:p50:HDAC-1 repressor complex orchestrates transcriptional inhibition of multiple pro-inflammatory genes[J]. J Hepatol, 2010, 53(3): 519-527.
[37] BIANCHI I, CARBONE M, LLEO A, et al. Genetics and epigenetics of primary biliary cirrhosis[J]. Semin Liver Dis, 2014, 34(3): 255-264.
[38] SELMI C, CAVACIOCCHI F, LLEO A, et al. Genome-wide analysis of DNA methylation, copy number variation, and gene expression in monozygotic twins discordant for primary biliary cirrhosis[J]. Front Immunol, 2014, 5: 128.
[39] HU Z, HUANG Y, LIU Y, et al. beta-Arrestin 1 modulates functions of autoimmune T cells from primary biliary cirrhosis patients[J]. J Clin Immunol, 2011, 31(3): 346-355.
[40] NINOMIYA M, KONDO Y, FUNAYAMA R, et al. Distinct microRNAs expression profile in primary biliary cirrhosis and evaluation of miR 505-3p and miR197-3p as novel biomarkers[J]. PLoS One, 2013, 8(6): e66086.
[41] WEBB GJ, SIMINOVITCH KA, HIRSCHFIELD GM. The immunogenetics of primary biliary cirrhosis: a comprehensive review[J]. J Autoimmun, 2015, 64: 42-52.
[42] CIRULLI ET, GOLDSTEIN DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing[J]. Nat Rev Genet, 2010, 11(6): 415-425.
[43] SUN L, ZHANG X, HE L. GWAS promotes precision medicine in China[J]. J Genet Genomics, 2016, 43(8): 477-479.
[44] DONG M, LI J, TANG R, et al. Multiple genetic variants associated with primary biliary cirrhosis in a Han Chinese population[J]. Clin Rev Allergy Immunol, 2015, 48(2-3): 316-321.
[45] LAMMERS WJ, HIRSCHFIELD GM, CORPECHOT C, et al. Development and validation of a scoring system to predict outcomes of patients with primary biliary cirrhosis receiving ursodeoxycholic acid therapy[J]. Gastroenterology, 2015, 149(7): 1804-1812.
[46] SAMUR S, KLEBANOFF M, BANKEN R, et al. Long-term clinical impact and cost-effectiveness of obeticholic acid for the treatment of primary biliary cholangitis[J]. Hepatology, 2017, 65(3): 920-928.
[47] HIRSCHFIELD GM, MASON A, LUKETIC V, et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid[J]. Gastroenterology, 2015, 148(4): 751-761.
[48] NEVENS F, ANDREONE P,MAZZELLA G, et al. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis[J]. N Engl J Med, 2016, 375(7): 631-643.
[49] HEGADE VS, KHANNA A, WALKER LJ, et al. Long-term fenofibrate treatment in primary biliary cholangitis improves biochemistry but not the UK-PBC risk score[J]. Dig Dis Sci, 2016, 61(10): 3037-3044.
[50] MOUSA H, LLEO A, INVERNIZZI P, et al. Advanced in pharmacotherapy for primary biliary cirrhosis[J]. Expert Opin Pharmacother, 2015, 16(5): 633-643.
[51] EGAWA H, SAKISAKA S, TERAMUKAI S, et al. Long-term outcomes of living-donor liver transplantation for primary biliary cirrhosis: a Japanese multicenter study[J]. Am J Transplant, 2016, 16(4): 1248-1257.
[52] WEBB CF, BRYANT J, POPOWSKI M, et al. The ARID family transcription factor bright is required for both hematopoietic stem cell and B lineage development[J]. Mol Cell Biol, 2011, 31(5): 1041-1053.
[53] MYERS RP, SWAIN MG, LEE SS, et al. B-cell depletion with rituximab in patients with primary biliary cirrhosis refractory to ursodeoxycholic acid[J]. Am J Gastroenterol, 2013, 108(6): 933-941.
[54] DHIRAPONG A, LLEO A, YANG GX, et al. B cell depletion therapy exacerbates murine primary biliary cirrhosis[J]. Hepatology, 2011, 53(2): 527-535.
[55] HIRSCHFIELD GM, GERSHWIN ME, STRAUSS R, et al. Ustekinumab for patients with primary biliary cholangitis who have an inadequate response to ursodeoxycholic acid: a proof-of-concept study[J]. Hepatology, 2016, 64(1): 189-199.
[56] TACKE F, LUEDDE T, TRAUTWEIN C. Inflammatory pathways in liver homeostasis and liver injury[J]. Clin Rev Allergy Immunol, 2009, 36(1): 4-12.
[57] XIAO L, JIA L, ZHANG Y, et al. Human IL-21+IFN-gamma+CD4+T cells in nasal polyps are regulated by IL-12[J]. Sci Rep, 2015, 5: 12781.
[58] MEAGHER C, BEILKE J, ARREAZA G, et al. Neutralization of interleukin-16 protects nonobese diabetic mice from autoimmune type 1 diabetes by a CCL4-dependent mechanism[J]. Diabetes, 2010, 59(11): 2862-2871.
引证本文:LI YK, MA X, TANG RQ. Advances in genetic research on primary biliary cholangitis[J]. J Clin Hepatol, 2017, 33(11): 2105-2111. (in Chinese)
李奕康, 马雄, 唐茹琦. 原发性胆汁性胆管炎的遗传学研究现状[J]. 临床肝胆病杂志, 2017, 33(11): 2105-2111.
(本文编辑:邢翔宇)
Advancesingeneticresearchonprimarybiliarycholangitis
LIYikang,MAXiong,TANGRuqi.
(DepartmentofGastroenterology,RenjiHospital,RenjiHospital,ShanghaiJiaoTongUniversitySchoolofMedicine&ShanghaiInstituteofDigestiveDiseases,Shanghai200001,China)
Primary biliary cholangitis (PBC) is an autoimmune liver disease with strong genetic susceptibility. The genome-wide association studies and immunochip studies conducted in recent years help to reveal the pathogenesis of PBC. The susceptibility genes of PBC are classified into human leukocyte antigen gene and non-human leukocyte antigen gene, and most of the susceptibility loci are associated with immune regulation, suggesting that disorders of the immune regulatory pathways may play an important role in the pathogenesis of PBC. In addition, the weighted genetic risk score of these candidate genes may predict the risk of PBC. At present, about one third of PBC patients have suboptimal response to ursodeoxycholic acid; therefore, targeted drugs for susceptibility genes may become an effective substitutive therapy.
cholangitis, biliary; cholestasis; genetic predisposition to disease
R575.22
A
1001-5256(2017)11-2105-07
10.3969/j.issn.1001-5256.2017.11.011
2017-08-24;
2017-09-29。
李奕康(1994-),男,主要从事自身免疫性肝病组学转化研究。
唐茹琦,电子信箱:ruqi_tang@126.com。
