纳米多孔金属电催化剂在氧还原反应中的应用
2017-11-01翟萧丁轶
翟 萧 丁 轶
(1上海理工大学材料科学与工程学院,上海 200093;2天津理工大学新能源材料与低碳技术研究院,材料科学与工程学院,天津 300384;3天津市先进多孔功能材料重点实验室,天津 300384)
纳米多孔金属电催化剂在氧还原反应中的应用
翟 萧1,3丁 轶2,3,*
(1上海理工大学材料科学与工程学院,上海 200093;2天津理工大学新能源材料与低碳技术研究院,材料科学与工程学院,天津 300384;3天津市先进多孔功能材料重点实验室,天津 300384)
燃料电池是将化学能直接转化为电能的能量转换装置,具有绿色、高效、便携等特点。对于大多数使用氧气或者空气为氧化剂的燃料电池而言,其阴极氧还原反应动力学缓慢、稳定性差是阻碍该技术走向商业化的主要因素,因此开发高催化活性和良好稳定性的低成本氧还原催化剂非常重要。基于脱合金法制得的纳米多孔金属是一类新型的宏观尺度纳米结构材料,其独特的开放型孔道结构、优良的导电性和结构的可调控性使其在电催化相关领域具有广泛的应用。本文侧重于讨论纳米多孔金属作为氧还原催化剂时所展示的一系列结构特性,及其在发展新一代高性能一体化燃料电池催化剂中所展示的机会。
纳米多孔金属;脱合金;燃料电池;氧还原;低铂催化剂
1 引 言
能源与生态环境可持续性发展已经成为当今世界的主题,而目前全球 80%的能耗来源是不可再生的化石燃料,化石燃料在使用过程中会释放CO2,从而引起气候变化和其他一系列环境问题。因此开发可再生清洁能源,研发高效能源转换、存贮装置是解决此类问题的主要手段。燃料电池作为本世纪备受关注的新能源技术之一,可实现化学能向电能的直接转换,具有高效、便携、环境友好等特点,且使用温度范围较广,目前已在可移动电源和城市公共交通等领域成功示范1−3。燃料电池技术目前面临的最大问题是由于技术链长、造价高昂、目标市场尚不明确等导致一直未能实现大规模商业应用。从材料角度看,寻找合适的材料,尤其是催化剂材料,并匹配相应的组装工艺,实现制造成本大幅度降低,同时提高其功率密度和系统稳定性是燃料电池的最核心技术。
19世纪60年代,科学家们即采用将粒径2−5 nm的铂(Pt)微晶负载在碳粉或者碳纸上的方式来降低Pt的载量2,这也许是最早的纳米结构工程,也是燃料电池催化剂发展的重要节点。但当前各类燃料电池仍然使用了过多的Pt催化剂,使得地球上Pt资源特别匮乏的问题额外突出。这个问题对于中国尤其明显,因为中国同时是Pt需求大国和Pt资源匮乏国,每年Pt产量不足4吨,90%以上依赖进口4。由于阴极氧还原反应机理复杂、反应动力学缓慢,阴极催化剂Pt载量可比阳极高8倍之多5,6,因此,近 10年来,开发高性能低 Pt或非Pt氧还原催化剂已成为燃料电池领域研究的热点。而脱合金法制备的纳米多孔金属具有可调控的双连续开放式孔结构、清洁表面、优良的导电性、制备工艺简单、可自支撑等特点,这为开发高效、低Pt或非Pt燃料电池催化剂提供了新的机会。
2 氧还原反应
2.1 氧还原机理
氧还原反应(ORR)是一个多电子反应过程,其中间产物较多且难以利用原位检测来进行分析。研究者们已提出多种ORR机理来分析O2在催化剂表面的还原过程,目前较为大家接受的解释金属表面ORR复杂过程的是Wroblowa机理7。以水溶液中金属表面的ORR为例,该机理认为O2在金属表面电化学还原时有两种可能路径,可以通过2电子途径生成中间产物H2O2,也可以通过4电子途径直接生成水。其中O2经由2电子途径生成的H2O2吸附在电极表面,可以继续进行电化学还原生成最终产物水,也可以在电极表面发生催化分解或者是发生解吸附溶解至溶液中。相较于2电子途径,4电子途径可有效地提高催化剂催化效率,并防止电极支撑材料和质子交换膜等材料被2电子途径产生的H2O2腐蚀。这两种反应途径都包括了O2分子的解离吸附以及与质子的反应,其中含氧物种在催化剂表面的吸附既是实现电子传递的必需条件,又是占据催化剂表面活性位点造成催化效率低下的原因8−11。在燃料电池应用中,ORR催化剂必须在高氧化电位即极易腐蚀的环境下保持高活性并兼具高稳定性:既要有足够的化学活性能够活化 O2(发生一个电子和一个质子的转移,生成吸附 OOH*),又要有适当的惰性使催化剂表面的产物 H2O成功解离12。因此了解 O2在催化剂表面的反应机理是提高ORR催化剂效率和稳定性的关键所在。但在实际的ORR反应过程中,其中间产物繁多(O*、OH*、OOH*等),且这些中间产物很难用原位分析方法检测到13,14,所以目前对ORR机理的认识并不充分。即使是已经得到广泛研究的贵金属 Pt基催化剂,其 ORR过程也尚未被完全揭示7,15−17。
近年来对ORR过程的研究越发深入,目前公认可通过调控以下参数来提高Pt催化剂的性能:1)缩短 Pt原子间距(几何效应);2)调控 d-带电子状态使d-带中心下移(电子结构);3)表面粗糙度。其中通过几何效应和电子结构的调控是通过减弱含氧物种在 Pt催化剂表面的吸附来提高其催化效率,而提高表面粗糙度可有效增加催化剂表面的活性位点,从而提高催化效率。


2.2 氧还原催化剂发展现状
燃料电池阴极反应主要面临以下问题:1)电压损失严重。这一问题主要是由于阴极ORR需要较高过电势,且阳极燃料分子经质子(或离子)交换膜渗透到阴极产生混合电位等引起;2)催化效率低。如前所述ORR实际的中间产物比较复杂,且这些中间产物既是电子传递的媒介,又是造成催化剂催化效率低下的主要原因。针对以上问题,研究者们研发了大量新型 ORR催化剂,例如 Pt/贵金属基、过渡金属化合物、生物材料、大环化合物和掺杂碳材料催化剂等18−23。从综合性能看,Pt族贵金属基催化剂目前仍然是最现实的可满足应用端对电源系统整体要求的材料19−21,24。
前文已提到可通过几何效应、电子结构和表面粗糙度来提高催化剂催化效率,而研究发现合金化可以有效改变催化剂的电子结构,结合材料的纳米化进而改变催化剂的表面应力和能带结构,可达到提高催化剂活性的目的。不同的纳米结构如纳米花25、微纳颗粒26、纳米颗粒27、纳米管28和核壳纳米结构29等都已被报道,其中核壳结构的催化剂由于其独特的结构优势可显著提高ORR催化剂的催化活性,同时可降低Pt含量,因而得到了广泛的关注。图 1是根据文献资料进一步归纳的目前常用的制备Pt-基核壳结构催化剂的方法30,由于内层原子与外层原子之间的配位效应和表面应力的协同作用,可使催化剂电子结构d-带中心下移31,32,相较于纯 Pt催化剂可减弱与中间产物间的吸附作用,从而提高其催化活性29,33。
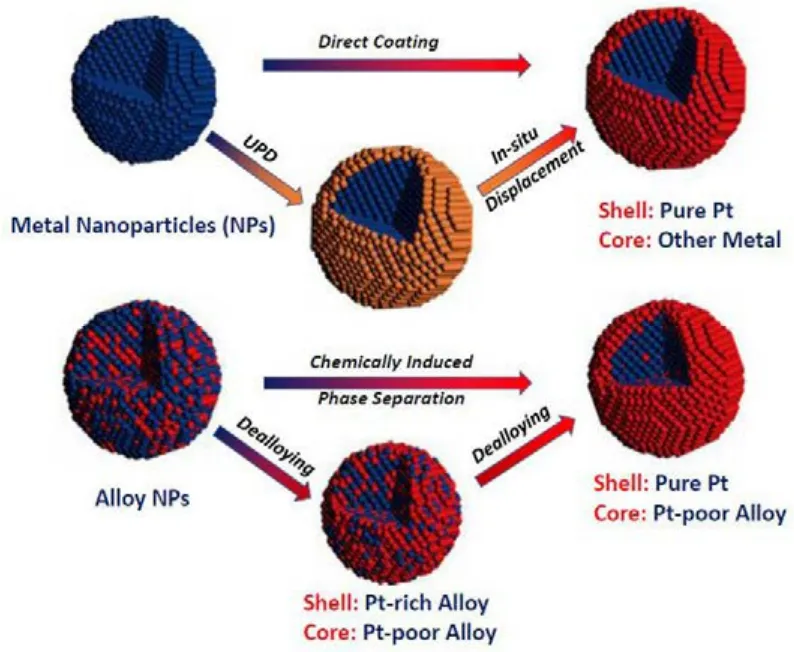
图1 典型的核壳结构Pt基纳米电催化剂制备方法30Fig.1 Typical methods for the synthesis of core-shell structured Pt-electrocatalysts30.
如果把纳米晶不同的形貌(团簇、球状颗粒、线状、管状、片状、枝晶等)考虑在一起,那么文献研究的大多数ORR催化剂均可视为颗粒型催化剂,而颗粒型催化剂在具体应用时需要与合适的催化剂载体复合。最常用的催化剂载体是高比表面碳材料,这种“先各自制备−后复合组装”的催化剂结构通常会遇到催化剂和载体的兼容性问题,这对于需要兼顾结构通透性、导电子性和导离子性的燃料电池膜电极来说尤其严重。很多 Pt颗粒催化剂难以在实际应用中发挥作用。有文献报道对于碳载铂(Pt/C)纳米催化剂结构,在燃料电池工况条件下,仅有不足30%的Pt在发挥作用34。此外,金属纳米颗粒与碳载体之间作用力通常较弱,在工况条件下,纳米催化剂易脱离载体材料发生团聚,这是目前颗粒型催化剂稳定性较差的主要原因。在这种情况下,发展具有三维立体结构的非颗粒型ORR催化剂变得十分有价值;而利用脱合金法制得的纳米多孔金属由于其三维贯穿的自支撑韧带孔道结构和优良的机械稳定性,并有望同时优化电子、离子和介质分子传递通道,是一类很有前景的结构型催化剂。
3 纳米多孔金属
纳米多孔金属是一种由纳米尺度的固体韧带(孔壁)和孔道在三维空间相互交织构成的海绵状的金属功能材料35。这类结构中的韧带和孔道在三维空间均可延伸,这实现了电催化所需的介质分子和电子的无阻碍传输,且由于其良好的导电性、高比表面积和特殊的具有凹/凸曲率的韧带结构,使其成为非常有前景的电极材料或构建复合电极所需的骨架材料,已在电化学能量存储和转换的应用中显示出巨大的潜力36−38。
3.1 纳米多孔金属的制备
纳米多孔金属的制备方法有很多种,如脱合金法35,模板法39,阳极氧化法40,激光蚀刻法41,燃烧合成法42,溶胶-凝胶法43,热分解法44等。在所有上述方法中,脱合金法由于原理简单、操作便捷、参数可调、适用性宽等特点而具有显著优势,图 2是典型的脱合金法制备纳米多孔金属的工艺流程图。脱合金是一种选择性蚀刻掉合金材料中的相对活泼元素,而剩余的相对不活泼元素自发重组形成纳米多孔骨架结构的电化学过程。为满足各种应用的需求,微观结构调控是纳米多孔金属材料开发的关键。可通过合金前驱体的设计,控制脱合金参数,材料后续热处理和表面改性等方法进行纳米多孔结构和功能的调控。
3.1.1 前驱体的选择
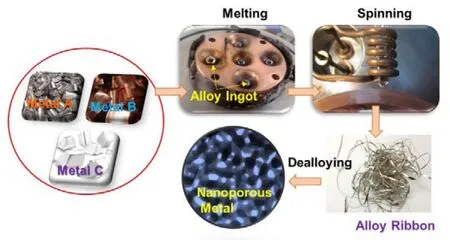
图2 脱合金法制备纳米多孔金属的典型过程Fig.2 Schematic illustration of the fabrication process of nanoporous metals by dealloying.
Erlebacher45提出,在脱合金过程中产生多孔结构的合金体系需具有以下基本特征:(i)合金组分中不同元素之间的电位差(溶解不同金属单质时所需电化学电位之间的差值)须达到几百毫伏;(ii)组分中通常含有比例较大的非贵金属组元(贵金属组元的含量低于某阈值);(iii)合金必须是均匀的,在溶解之前不存在相分离,即多孔结构在溶解过程中动态形成,而不是简单两相材料发生了单相析出和溶解;(iv)贵金属原子在合金/电解质界面处的扩散必须足够快。基于上述脱合金前驱体选择标准,许多合金体系已被探索,如 Al基46、Zn基47、Mg基48、Mn基49、Cu基50、Ni基51合金等。当然这些特征更多是经验性的,针对不同的合金及腐蚀介质均有较宽的可调控范围。
3.1.2 脱合金参数控制
脱合金过程中的一些参数包括电解质溶液的种类、浓度、腐蚀温度、施加电位、反应持续时间等都对纳米多孔金属产物的结构形貌有影响。首先是脱合金方式的选择,化学或电化学脱合金都可以获得纳米多孔结构,但是这两种方法得到的纳米多孔金属具有不同的特征尺寸,甚至具有不同的微观结构52−54。脱合金也可以在非水溶液(有机电解质、金属熔体或超临界流体)中进行,利用不同浓度腐蚀液制备的纳米多孔结构其孔径大小也不同。此外温度也是影响产物结构的重要因素,低温脱合金可以有效控制纳米多孔金属的孔径尺寸。一般来说,温度越低,贵金属元素的表面扩散速率就越慢,因此有助于获得韧带孔道尺寸较小的纳米多孔金属。而较高的温度会使纳米多孔金属结构粗化,但由于应力释放也有助于减少产物结构中的裂纹,可改善产物结构均一性和完整性。在电化学脱合金中电势的选择和施加方法对纳米多孔结构的形成也有显著影响。通常为了使非贵金属元素溶解和纳米多孔结构的演变能够发生,所施加的电势应当高于其临界电势但低于贵金属元素的平衡腐蚀电势。
结合不同的脱合金条件是调控纳米多孔结构的有效方法,例如Weissmüller等55报道了通过两步电化学脱合金法制备具有多级孔结构的纳米多孔金:第一步脱合金生成作为中间产物的纳米多孔 AuAg合金,将粗化后的合金韧带作为二次腐蚀的前驱体,可生成二级孔结构。此外,越来越多的研究表明气氛对多孔结构的形成也有一定影响。如Strasser等56研究了腐蚀气氛(空气、N2)和颗粒尺寸对 PtNi3纳米颗粒化学脱合金结构的影响。他们发现纳米孔的形成演化与在空气条件下Ni的剧烈溶解有关;相比之下,在N2保护下的无氧酸中脱合金则可抑制多孔结构的演化,从而形成富Pt表面的固体核壳颗粒结构。为了进一步调控目标产物的形态结构,经常对纳米多孔金属进行退火等后处理,该过程可以诱导多孔结构粗化从而得到特定尺寸维度的功能材料57。
3.2 纳米多孔金属的结构特性
众所周知,纳米材料与纳米结构的制备方法通常分为“从上而下(Top-down)”的材料刻蚀工艺和“自下而上(Bottom-up)”的材料形核生长工艺两大类。而纳米多孔金属材料的制备过程则有效结合了这两类工艺的基本特征,在于其表面上看是采用了Top-down类型的材料溶蚀工艺,而其微观组织结构的形成与演化却完全是个 Bottom-up过程。在脱合金过程中,越来越多的相对惰性金属原子被释放出来,它们会在原有固液界面上以类似于调幅分解(spinodal decomposition)的机制发生动态相分离,其中的固态相在亚纳米尺度形核生长58。因此,从结构几何看,纳米多孔金属的双连续结构和冶金学中的合金相变以及嵌段共聚物相分离产物的形貌极其相似59,60。此外,由于纳米多孔金属表面原子因配位不饱和而具有较高的表面扩散速率,因此可以利用这个特征对其进行后续的结构调控。如图 3(a−c)所示,纳米多孔金属的韧带孔道尺寸可以在低至2−3 nm,大到数个微米的宽范围内进行调控。
3.2.1 纳米多孔金属的微观结构
由于纳米多孔金属的微观组织结构通常要比原始合金前驱体的晶粒尺寸小 2−3个数量级,其显微结构在透射电子显微镜表征时经常体现出多孔单晶特征(如图3(d−f))。同时由于脱合金过程大都可以在无机电解质溶液中进行,无需有机或者高分子表面修饰试剂的参与,因此纳米多孔金属通常具有较为洁净的表面结构。这些结构特征有助于纳米多孔金属电极的高导电性和高表面反应活性。此外,这类宏观尺度纳米结构材料也拥有良好的机械稳定性和导电导热性能64,65。电学性能测试显示,纳米多孔金电极的导电性能在1 × 10−6Ω·m 量级,与传统碳黑或多孔碳电极相比,其电阻率至少降低 4个数量级以上,这对于后期研发高功率燃料电池电堆尤其重要。

图3 (a−c) 具有不同特征尺寸的纳米多孔金的SEM图像; 纳米多孔金的(d) TEM图像61; (e−f) HRTEM图像62,63Fig.3 (a−c) SEM images of nanoporous gold (NPG) with different pore sizes; (d) TEM61 and(e−f) HRTEM image62,63 of NPG.
传统的以模板法、溶胶-凝胶法、热分解法等手段制得的多孔结构材料,从微观尺度分析均可以理解为微纳米颗粒的定向组装,因此其本征表面特性基本上与其组装单元微纳米颗粒相似。而基于脱合金方法制得的纳米多孔金属则没有类似的组装单元。早在2007和2008年,Weissmüller68和 Chen64,66课题组独立地实施了透射电子显微镜断层扫描技术进而实现了纳米多孔金的形貌三维重构(如图 4a所示)。定量的数据分析显示,纳米多孔金孔道和韧带在拓扑学和形态学上完全等价。此外,这样一个随机均匀的双连续结构具有一定的准周期(quasiperiodic)特性,韧带和孔道的界面类似于双曲面,正负曲率相当,总表面曲率接近于零(如图 4b所示),从几何上看像一个螺旋形的gyroid结构69。最近,Chen课题组67利用球差矫正扫描透射电子显微镜对纳米多孔金的韧带结构实施了离散断层扫描分析,率先实现了纳米多孔金韧带结构的原子分辨三维重构(如图 4(c−e)所示)。研究一方面进一步证实了纳米多孔金表面结构的双曲率几何,也揭示了该多孔结构与传统的低维纳米颗粒如截角八面体金颗粒(TOP)在表面低配位原子分布方面具有本质的不同。如图 4e所示,对于平均韧带/直径尺寸相当的NPG和TOP而言,低配位数(CN = 5−8)所占表面原子的比例在NPG结构中要远远大于TOP。例如,韧带尺寸在10.3 nm的NPG,其表面低配位数原子比例仍高达42.1%,显著高于直径在3.4 nm的金颗粒所能体现的38.1%。由于材料表面配位不饱和的原子通常具有更高的反应活性,也是各催化反应活性中心的主要位点,这一方面能够佐证纳米多孔金属在非均相催化反应中已经体现出的诸多异常低温反应特性70,也预示着它们在电催化方面也将有新的机会。
3.2.2 纳米多孔金属的宏观结构
由于合金前驱体来源丰富,形貌多样,而脱合金过程也有其调控的灵活性,因此纳米多孔金属可完美结合金属材料良好的机械性能和纳米材料优异的界面特性,这大大拓宽了纳米多孔金属的应用范围。通过对材料组分和脱合金参数的合理控制,具有不同微观组织结构的纳米多孔金属都可以被制备出来,从而适应于不同的应用环境。如图5所示,纳米多孔金属可以是粉末、条带71、薄膜72,73,也可以是厚度达到几厘米的块体46,这种宏观特性不仅为纳米多孔金属的结构多样性提供了更多的可能,也拓宽了纳米多孔金属的应用范围。
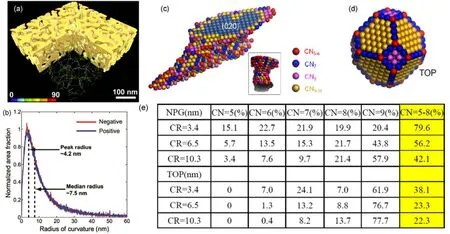
图4 (a)纳米多孔金的电子断层扫描图像64;(b)归一化到面积后的韧带曲率半径分布66;(c)纳米多孔金韧带的原子分辨三维重构图;(d)金纳米颗粒的八面体示意图;(e)不同尺寸的纳米多孔金和截角八面体金颗粒中表面不同配位数(5−9)的原子所占表面原子的比例67Fig.4 (a) 3D skeletal network of NPG imaged by electron tomography64; (b) Normalized area fractions plotted against
4 纳米多孔金属氧还原催化剂
如前所述,ORR催化剂表面电子结构和配位效应可影响催化剂与吸附物种间结合能大小,并进而决定催化剂综合活性。Norskov等74利用密度泛函理论揭示了不同金属与其ORR催化性能之间的火山型关系,其中金属Pt是最好的单金属ORR催化剂,这与实验结果完全一致。根据这一火山型关系,合金化可用于调节(降低)Pt-O结合能,而脱合金制备技术可有效兼顾产物高比表面积和微观组织结构,因此,近年来利用脱合金法制备ORR催化剂的文献大量涌现。下面根据已有报道概述一下纳米多孔 ORR催化剂近年来取得的主要进展。

图5 (a−c)各种类型的合金前驱体;(d−e)相应的宏观尺度纳米多孔金属46,73Fig.5 (a−c) Various alloy precursors; (d−e) the resulted nanoporous metal membranes and chunks46,73.
4.1 多孔纳米颗粒
通过在Pt系贵金属表层和次表层掺杂非贵金属元素31,或是在非贵金属表面修饰Pt是获得高性能ORR催化剂的有效途径75。2007年,Strasser76报道了可显著提高商业化碳载铂(Pt/C)催化剂ORR性能的简单方法:将Pt/C在硝酸铜溶液中浸渍,通过高温退火(600−950 °C)实现铜盐分解并同时与Pt合金化生成PtCu双金属纳米粒子。对所得材料进行电化学脱合金,发现随着铜的溶出,产物的ORR逐步提高。结构分析显示,所获得的电催化剂内核为 Pt79Cu21,表层为Pt93Cu7,其ORR活性比未处理商业Pt/C提高4−6倍。该课题组随后采用相似的方法对 Pt/C表面修饰铜钴两种金属,通过对所获得的三元 PtCuCo进行脱合金处理,所制得阴极催化剂的ORR性能也有显著提高,并在初步的膜电极测试中得到进一步验证77。由于催化单元尺寸较小,这些报道未关注脱合金过程中是否有孔结构形成,晶格收缩和表面微结构的变化被认为是ORR性能提高的关键。
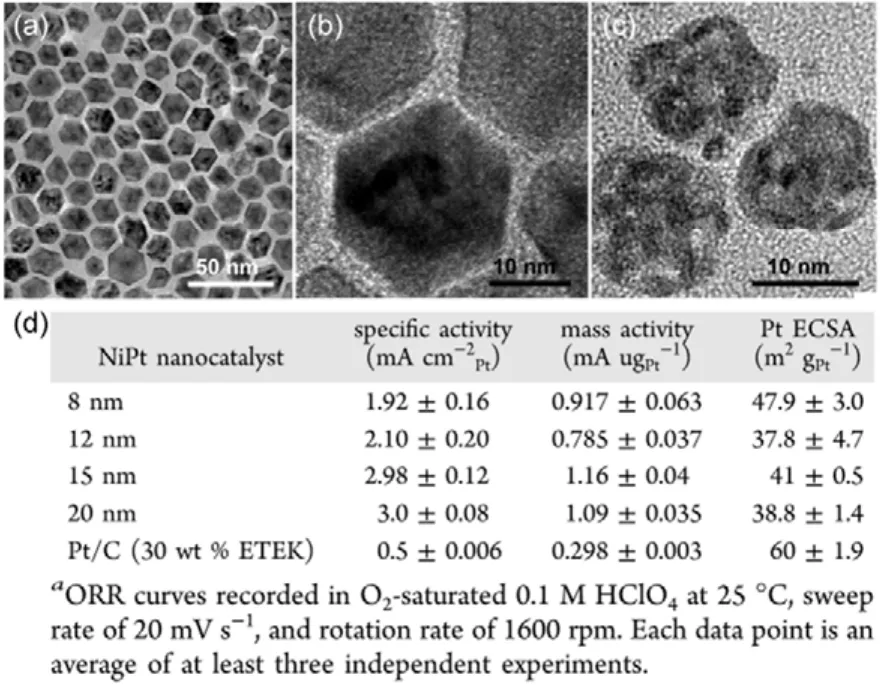
图6 Pt-Ni纳米颗粒的(a)TEM图像和(b)HRTEM图像;(c)纳米多孔PtNi合金的HRTEM图像78;(d)脱合金法制备的纳米多孔PtNi纳米颗粒和Pt/C(30% (w))催化剂的性能对比81Fig.6 (a) TEM and (b) HRTEM images of PtNi alloy nanoparticles, (c) HRTEM image of nanoporous PtNi alloy nanoparticles78; (d) iR-Free ORR kinetic activity parameters for dealloyed PtNi catalysts and 30% (w) Pt/C81.
有关Pt-基纳米多孔纳米颗粒的制备最早见于2011年。Li等78发展了化学合成技术制备了单分散双金属合金纳米颗粒,然后用脱合金方法去除其中的活性组分。电子显微分析清楚地证明了多孔结构纳米粒子的形成(如图 6(a−c)所示)。随后Oezaslan等79研究了脱合金法制备的二元 Pt-Co和 Pt-Cu合金纳米颗粒催化剂的形貌结构和粒内成分与前驱体粒径大小间的关系,他们发现当前驱体粒径小于20 nm时,便无法形成孔结构。而当前驱体粒径大于30 nm时,脱合金后的Pt-基合金则倾向于形成纳米级的多孔结构。相似的方法后来被Erlebacher等80,81采纳去研究纳米多孔PtNi纳米颗粒的结构演化及其ORR构效关系。通过对前驱体合金纳米颗粒尺寸和组分的探讨,他们同样发现只有当合金颗粒粒径大于15 nm时,多孔结构才能形成。所制得的纳米多孔PtNi纳米颗粒的韧带尺寸小低至2 nm,比表面积比商业Pt/C略低,但总体Pt质量ORR活性在0.9 V (vs RHE)条件下比Pt/C高约4倍。
利用定位TEM电镜技术,Baldizzone等82深入研究了纳米多孔PtNi纳米颗粒的电化学失活机理。通过模拟燃料电池真实工况条件实施的加速老化实验,发现该多孔阴极催化剂在0.4−1.0 V (vs RHE),区间结构和电化学活性比较稳定,但当电位区间扩展到0.4−1.4 V (vs RHE)时,多孔结构显著粗化并导致失活。该工作显示多孔结构ORR催化剂在迈向应用时仍然需要进一步改善其结构稳定性。
4.2 Pt-基纳米多孔合金
2010年,Erlebacher等94报道了用离子液体改善阴极催化剂ORR性能的新思路。他们首先利用电化学脱合金方法在NiSO4溶液中腐蚀Pt23Ni77合金条带,然后在所获得的纳米多孔PtNi合金上修饰微量的疏水性全氟乙基磺酰基酰亚胺阴离子基离子液体[MTBD][beti]。这类质子型离子液体具有疏水性、良好的溶氧能力和质子导电性,因此可以大幅度改善PtNi合金的ORR性能。在0.9 V(vs RHE)条件下测算的本征动力学电流密度值约18.2 mA·cm−2,与理想的 Pt3Ni(111)单晶的本征活性相当,比商业Pt/C几乎提高了两个数量级。相似的方法后来又被该课题组应用于修饰纳米多孔纳米颗粒阴极催化剂,同样也获得了很好的性能 83−85。
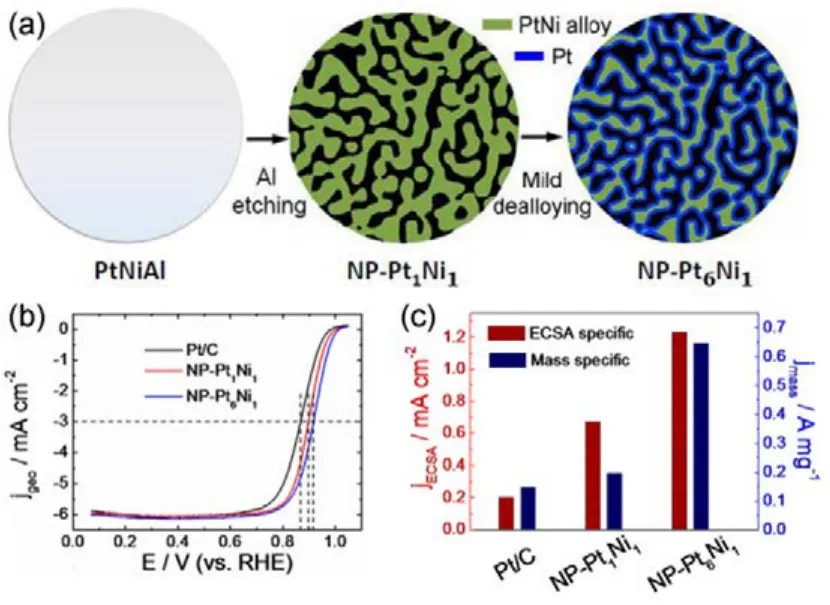
图7 (a)脱合金法制备纳米多孔PtNi合金催化剂示意图;(b)纳米多孔PtNi合金催化剂和Pt/C催化剂的极化曲线图;(c) 0.9 V (vs RHE)处质量活性和面积活性对比图86Fig.7 (a) Schematic illustration of the fabrication process of nanoporous PtNi surface alloy catalyst;(b) ORR polarization curves and (c) ECSA and Pt mass specific kinetic current densities at 0.9 V(vs RHE) for nanoporous PtNi and Pt/C86.
2012年,Ding等86报道了制备核壳结构纳米多孔合金阴极催化剂的通用方法。他们首先使用NaOH溶液从组分明确的PtNiAl三元合金前驱体中浸出较活泼的组分Al,制备出纳米多孔PtNi合金;然后使用HNO3溶液进一步浸出PtNi韧带表层的组分Ni,最后可获得表层纯铂内核为PtNi合金的核壳结构纳米多孔 PtNi表面合金阴极催化剂。他们比较了所获得的表观组分为 Pt1Ni1和Pt6Ni1的两种电催化剂的ORR性能,如图7所示,在相同实验条件下其半波电位分别为 0.897和0.916 V,分别比商业Pt/C催化剂(0.866 V)高出31和50 mV。尤其是核壳结构纳米多孔Pt6Ni1合金,其 0.9 V (vs RHE)处 Pt质量活性可达到 0.65 A·mg−1,比Pt/C提高5倍以上。此外这类催化剂还表现出比 Pt/C更好的电化学稳定性。Pt和 Ni之间的合金化效应和材料表面应变(压缩)效应被认为是这类材料具有较高ORR本征活性的原因;而其开放的纳米孔道结构也便于物质传输,有助于电极反应动力学过程。
相似的方法后来被 Xu87−90和 Zhang91−93等课题组发展应用于制备其他纳米多孔PtM (M = Pd,Cu, Fe, Ti,… )合金阴极催化剂。其反应前驱体通常是PtMX三元乃至更多元合金,其中X是比Pt和M更活泼的金属元素,例如 Al、Mg等。通过调控Pt/M之间的比例以及X的溶出过程,可以有效调控纳米多孔合金产物的比表面积、表面组分和结构,进而达到增强其ORR催化活性和稳定性的目的。
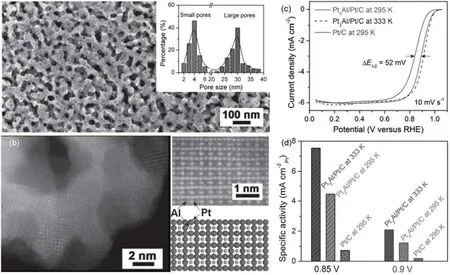
图8 (a)纳米多孔Pt3Al/Pt的SEM图像和孔径分布图(内嵌图);(b) 纳米多孔Pt3Al/Pt的HAADF-STEM图、超晶格放大图和相应原子结构模型;(c,d)纳米多孔Pt3Al/Pt/C和Pt/C的ORR极化曲线和比面积活性对比图Fig.8 (a) SEM image of Pt3Al/Pt and pore size distribution (inset); (b) HAADF-STEM images of Pt3Al with super lattice feature and simulated atomic model; (c) ORR polarization curves of Pt3Al/Pt/C and Pt/C; (d) Comparison of specific kinetic activity for Pt3Al/Pt/C and Pt/C.
2014年,Jiang课题组94以Pt12Al88(原子百分比)合金条带为前驱体,1 mol·L−1NaOH溶液为电解质,氮气保护下脱合金制得了具有双级孔分布的纳米多孔PtAl合金ORR催化剂。如图8所示,整体材料呈现典型的双连续纳米多孔结构,此外,在30 nm直径的韧带上广泛分布有约4 nm的孔洞。精细的球差电镜分析显示,材料体相是Pt3Al或Pt5Al结构的金属间化合物,表层覆盖有原子级别厚度的纯铂。基于实验结果,他们实施了密度泛函理论(DFT)模拟,证实受内部压缩应力和表面配位效应的影响,表层Pt电子结构d带中心发生下移,可有效削弱Pt与氧化物种之间的结合,从而改善其ORR活性。电化学测试也确实证明,所制得材料电化学比活性在 0.9 V (vs RHE)比商业Pt/C提高5−6.3倍。尤其重要的是,该材料还具有优异的电化学稳定性和高温活性。在氮气保护的0.1 mol·L−1HClO4溶液中,以 50 mV·s−1的速率在0.6−1.1 V (vs RHE)电位区间循环40000圈之后,商业Pt/C催化剂半波电位发生了75 mV的负移,而纳米多孔PtAl合金催化剂只发生了约20 mV的负移。
4.3 非Pt纳米多孔金属
由于Pt资源稀缺且价格昂贵,近年来也有大量的研究致力于开发新型非Pt催化剂。以掺杂碳材料为代表的非Pt催化剂是该方向的研究热点,但其总体催化活性与 Pt族金属相比仍有显著差距,尤其是在酸性体系中14。Erlebacher等95曾研究脱合金腐蚀 AuAg合金得到的纳米多孔金的ORR活性,发现纳米多孔金可通过两个2电子反应逐步将O2还原为H2O。尤其是其第二步从H2O2还原到H2O的过程非常迅速,并且随着材料韧带尺寸的减小,其H2O2催化还原活性提高。他们认为多孔结构有效增加了 Au表面的低配位数反应活性位点,使得反应顺利进行。当然,总体而言纳米多孔金的 ORR催化活性与 Pt相比仍然相差甚远。在各类金属元素中,只有Pd的ORR活性比较接近Pt96,且相对更便宜。但Pd的化学稳定性较差,需要通过与其它组分形成复合结构以改善其综合电化学性能。
2009年,Ding等报道了利用CuAl脱合金制得的纳米多孔铜(NPC)作为模板和还原剂如H2PtCl6,K2PdCl4通过简单的置换反应,制备出具有三连续多级结构的PtCu和PdCu双金属电催化剂97,98。在置换反应中,贵金属元素不断沉积在纳米多孔铜韧带表面,而内层Cu原子不断被消耗,最终形成中空管状结构。所制得电催化剂总体呈现三维纳米多孔结构,而其韧带则是合金纳米管,管直径约60 nm,而壳层厚度在4−10 nm之间,并由更细小的合金纳米颗粒组成。在 0.1 mol·L−1HClO4溶液中,其ORR极化曲线半波电位为0.84 V,显著优于商业Pd/C催化剂的0.78 V,与商业Pt/C相当97。利用第4.2节中提及的脱合金方法对含Pd多元合金进行腐蚀处理,也可以简便地获得结构组分可控的纳米多孔 Pd基合金,如PdCu97,99,100,PdFe90,PdCo101−103,PdNi102,和PdTi103等。尤其是基于PdTiAl脱合金制得的纳米多孔PdTi合金,实验测得其ORR活性和稳定性均比Pt/C好,尤其是其对甲醇氧化几乎没有活性,因此是非常有希望的直接醇燃料电池阴极催化剂。
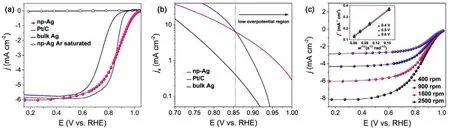
图9 纳米多孔银(np-Ag),Pt/C和块体Ag的(a) ORR极化曲线和(b) Tafel 曲线;(c) np-Ag循环5000圈前后的ORR极化曲线和相应的Tafel曲线(内嵌图)104Fig.9 (a) ORR polarization curves and (b) Tafel plots of np-Ag, Pt/C, and bulk Ag; (c) ORR curves and corresponding Tafel plots of np-Ag before and after 5000 potential (inset)104.

图10 基于脱合金方法可实现复杂结构多孔电极的设计与合成。(a)多级孔;(b)层次孔;(c)梯度孔;(d)多孔纳米管71,106;(e)多孔纳米胶囊;(f,g)多孔纳米棒及阵列结构107Fig.10 Dealloying allows customized design and fabrication of various hierarchically porous electrode materials,such as (a) multimodal porosity; (b) layer structure; (c) gradient porosity; (d) porous nanotubes71,106;(e) porous nanocapsules; (f,g) porous nanowires and arrays107.
近年来碱性燃料电池研究的复苏也吸引了相应高性能ORR催化剂的研发。由于碱性条件下许多电极材料都拥有良好的电催化活性,因此开发以Pt、Pd、Au、Ru等贵金属材料为主的阴极催化剂价值相对有限。相比之下,虽同属贵金属,银的经济适用性要强很多。Jiao等104研究了经AgAl脱合金制得的纳米多孔 Ag105在碱性条件下的ORR性能,发现其与Pt/C和体相Ag相比具有更低过电势,并通过计算电子转移数确认为 4电子ORR反应。他们推测纳米多孔 Ag结构内部和外部的Ag原子可能具有不同的电化学行为,但未解释原因。其中一个可能的解释是孔道内部Ag原子与表面Ag原子具有不同配位数和空间环境,因而具有不同的反应活性。如图9所示,纳米多孔Ag阴极催化剂在低过电势区具有更大的电流密度,且在循环5000圈后几乎没有电流损失。这表明纳米多孔Ag在碱液中具有良好的ORR催化活性和稳定性。
5 结论与展望
纳米多孔金属材料经过十多年的发展已经取得了长足的进步,其合金原材料来源丰富,制备过程简单灵活,产物结构可控且易于宏量制备。通过与其它材料制备工艺相结合,纳米多孔金属电极材料可以实现功能导向的设计。例如,图 10展示了基于脱合金制备技术可获得的各类复杂的多孔电极结构,包括了多级孔、层次孔、梯度孔、多孔纳米管、多孔纳米胶囊、多孔纳米棒及阵列结构。各功能单元的尺寸及组分可以相对独立调控,因此理论上有望调和现有燃料电池膜电极结构中的三相界面困境,即催化活性和稳定性的匹配、电子导电性和离子导电性的匹配、高通透性和高体积密度的匹配、和各电池部件之间亲疏水的匹配等。
也应该看到,纳米多孔金属材料的研发整体上还处于积累数据和探索可行性的“试错”阶段。以燃料电池催化为例,大部分的研究尚未针对膜电极现有问题来开展。许多材料一方面与现有膜电极工艺不兼容,另外受限于其结构,难以在实际电池中进行性能评估。如何理性设计并精准合成有明确功能的电极材料,首先要理解膜电极乃至系统层面上电子、质子传递和物料输运之间的协调,以及各反应界面稳定性,而这有待于材料学、电化学和电池工程各相关领域的同行通力协作。这既是学术领域的发展趋势,也是相关产业革新的必由之路。
(1) Brandon, N. P.; Skinner, S.; Steele, B. C. H. Annu. Rev. Mater. Res.2003, 33 (1), 183. doi: 10.1146/annurev.matsci.33.022802.094122
(2) Steele, B. C. H.; Heinzel, A. Nature 2001, 414 (6861), 345.doi: 10.1038/35104620
(3) Carrette, L.; Friedrich, K. A.; Stimming, U. ChemPhysChem 2000, 1(4), 162. doi:10.1002/1439-7641(20001215)1:4<162::AID-CPHC162>3.0.CO;2-Z
(4) http://www.chinabaike.com/z/keji/ck/528495.html (accessed Dec 28,2016).
(5) Wagner, F. T.; Lakshmanan, B.; Mathias, M. F. J. Phys. Chem. Lett.2010, 1 (14), 2204. doi: 10.1021/jz100553m
(6) Bondarenko, A. S.; Stephens, I. E. L.; Hansen, H. A.; Perez-Alonso, F.J.; Tripkovic, V.; Johansson, T. P.; Rossmeisl, J.; Norskov, J. K.;Chorkendorff, I. Langmuir 2011, 27 (5), 2058. doi: 10.1021/la1042475
(7) Markovic, N. M.; Schmidt, T. J.; Stamenkovic, V.; Ross, P. N. Fuel Cells 2001, 1 (2), 105. doi: 10.1002/1615-6854(200107)1:23.0.CO;2-9
(8) Xia, Z. H.; An, L.; Chen, P. K.; Xia, D. G. Adv. Eng. Mater. 2016, 6(17), 1600458. doi: 10.1002/aenm.201600458
(9) Xia, W.; Mahmood, A.; Liang, Z. B.; Zou, R. Q.; Guo, S. J. Angew.Chem. Int. Ed. 2016, 55 (8), 2650. doi: 10.1002/anie.201504830
(10) Raj, C. R.; Samanta, A.; Noh, S. H.; Mondal, S.; Okajima, T.; Ohsaka,T. J. Mater. Chem. A 2016, 4 (29), 11156. doi: 10.1039/c6ta03300h
(11) Liu, Y. Y.; Yue, X. P.; Li, K. X.; Qiao, J. L.; Wilkinson, D. P.; Zhang, J.J. Coord. Chem. Rev. 2016, 315, 153. doi: 10.1016/j.ccr.2016.02.002
(12) Greeley, J.; Stephens, I. E. L.; Bondarenko, A. S.; Johansson, T. P.;Hansen, H. A.; Jaramillo, T. F.; Rossmeisl, J.; Chorkendorff, I.;Norskov, J. K. Nat. Chem. 2009, 1 (7), 552. doi: 10.1038/nchem.367
(13) Gewirth, A. A.; Thorum, M. S. Inorg. Chem. 2010, 49 (8), 3557.doi: 10.1021/ic9022486
(14) Gasteiger, H. A.; Kocha, S. S.; Sompalli, B.; Wagner, F. T. Appl. Catal.,B 2005, 56 (1−2), 9. doi: 10.1016/j.apcatb.2004.06.021
(15) Stephens, I. E. L.; Bondarenko, A. S.; Gronbjerg, U.; Rossmeisl, J.;Chorkendorff, I. Energy Environ. Sci. 2012, 5 (5), 6744.doi: 10.1039/c2ee03590a
(16) Zhu, C. Z.; Li, H.; Fu, S. F.; Du, D.; Lin, Y. H. Chem. Soc. Rev. 2016,45 (3), 517. doi: 10.1039/c5cs00670h
(17) Zhou, M.; Wang, H. L.; Guo, S. J. Chem. Soc. Rev. 2016, 45 (5), 1273.doi: 10.1039/c5cs00414d
(18) Gao, M. R.; Jiang, J.; Yu, S. H. Small 2012, 8 (1), 13.doi: 10.1002/smll.201101573
(19) Shahgaldi, S.; Hamelin, J. Carbon 2015, 94, 705. doi:10.1016/j.carbon.2015.07.055
(20) Sakaushi, K.; Antonietti, M. Acc. Chem. Res. 2015, 48 (6), 1591.doi: 10.1021/acs.accounts.5b00010
(21) Nie, Y.; Li, L.; Wei, Z. D. Chem. Soc. Rev. 2015, 44 (8), 2168.doi: 10.1039/c4cs00484a
(22) Bashyam, R.; Zelenay, P. Nature 2006, 443 (7107), 63. doi:10.1038/nature05118
(23) Peng, S.; Guo, H. L.; Kang, X. F. Acta Phys. -Chim. Sin. 2014, 30,1778. [彭三, 郭慧林, 亢晓峰. 物理化学学报, 2014, 30, 1778.]doi: 10.3866/PKU.WHXB201407112
(24) Stoerzinger, K. A.; Risch, M.; Han, B. H.; Shao-Horn, Y. ACS Catal.2015, 5 (10), 6021. doi: 10.1021/acscatal.5b01444
(25) Sun, S. H.; Yang, D. Q.; Villers, D.; Zhang, G. X.; Sacher, E.; Dodelet,J. P. Adv. Mater. 2008, 20 (3), 571. doi: 10.1002/adma.200701408
(26) Cheng, F. Y.; Su, Y.; Liang, J.; Tao, Z. L.; Chen, J. Chem. Mater. 2010,22 (3), 898. doi: 10.1021/cm901698s
(27) Sun, J. Z.; Shi, J.; Xu, J. L.; Chen, X. T.; Zhang, Z. H.; Peng, Z. Q.J. Power Sources 2015, 279 (17), 334. doi:10.1016/j.jpowsour.2015.01.025
(28) Chen, Z.; Waje, M.; Li, W.; Yan, Y. Angew. Chem. Int. Ed. 2007, 46(22), 4060. doi: 10.1002/anie.200700894
(29) Chen, C.; Kang, Y. J.; Huo, Z. Y.; Zhu, Z. W.; Huang, W. Y.; Xin, H. L.L.; Snyder, J. D.; Li, D. G.; Herron, J. A.; Mavrikakis, M.; Chi, M. F.;More, K. L.; Li, Y. D.; Markovic, N. M.; Somorjai, G. A.; Yang, P. D.;Stamenkovic, V. R. Science 2014, 343 (6177), 1339.doi: 10.1126/science.1249061
(30) Yang, H. Angew. Chem. Int. Ed. 2011, 50 (12), 2674.doi: 10.1002/anie.201005868
(31) Stamenkovic, V.; Mun, B. S.; Mayrhofer, K. J. J.; Ross, P. N.;Markovic, N. M.; Rossmeisl, J.; Greeley, J.; Norskov, J. K. Angew.Chem. Int. Ed. 2006, 45 (18), 2897. doi: 10.1002/anie.200504386
(32) Paulus, U. A.; Wokaun, A.; Scherer, G. G.; Schmidt, T. J.;Stamenkovic, V.; Markovic, N. M.; Ross, P. N. Electrochim. Acta 2002,47 (22−23), 3787. doi: 10.1016/s0013-4686(02)00349-3
(33) Yang, R. Z.; Leisch, J.; Strasser, P.; Toney, M. F. Chem. Mater. 2010, 22(16), 4712. doi: 10.1021/cm101090p
(34) Wang, C.; Waje, M.; Wang, X.; Tang, J. M.; Haddon, R. C.; Yan, Y. S.Nano Lett. 2004, 4 (2), 345. doi: 10.1021/nl034952p
(35) Erlebacher, J.; Aziz, M. J.; Karma, A.; Dimitrov, N.; Sieradzki, K.Nature 2001, 410 (6827), 450. doi: 10.1038/35068529
(36) Meng, F. H.; Ding, Y. Adv. Mater. 2011, 23 (35), 4098. doi:10.1002/adma.201101678
(37) Wang, R.; Liu, J.; Liu, P.; Bi, X.; Yan, X.; Wang, W.; Ge, X.; Chen, M.;Ding, Y. Chem. Sci. 2014, 5 (1), 403. doi: 10.1039/c3sc52792a
(38) Wang, R.; Liu, J.; Liu, P.; Bi, X.; Yan, X.; Wang, W.; Meng, Y.; Ge, X.;Chen, M.; Ding, Y. Nano Research 2014, 7 (11), 1569. doi:10.1007/s12274-014-0517-9
(39) Xiong, W. H.; Zhang, W. C.; Yu, C. P.; Shen, R. Q.; Cheng, J.; Ye, J. H.;Qin, Z. C. Acta Phys. -Chim. Sin. 2016, 32, 2093. [熊文慧, 张文超,俞春培, 沈瑞琪, 程 佳, 叶家海, 秦志春. 物理化学学报, 2016,32, 2093.] doi: 10.3866/ PKU.WHXB201604082
(40) Nishio, K.; Masuda, H. Angew. Chem. Int. Ed. 2011, 50 (7), 1603.doi: 10.1002/anie.201005700
(41) Naeth, O.; Stephen, A.; Roesler, J.; Vollertsen, F. J. Mater. Process.Technol. 2009, 209 (10), 4739. doi: 10.1016/j.jmatprotec.2008.11.042
(42) Tappan, B. C.; Steiner, S. A.; Luther, E. P. Angew. Chem. Int. Ed. 2010,49 (27), 4544. doi: 10.1002/anie.200902994
(43) Guo, X. Z.; Ding, L.; Yu, H.; Shan, J. Q.; Yang, H. Acta Phys. -Chim.Sin. 2016, 32, 1727. [郭兴忠, 丁 力, 于 欢, 单加琪, 杨 辉. 物理化学学报, 2016, 32, 1727.] doi: 10.3866/PKU.WHXB201604082
(44) Nie, L. H.; Tan, Q.; Zhu, W.; Wei, Q.; Lin, Z. K. Acta Phys. -Chim. Sin.2015, 31, 1815. [聂龙辉, 谭 侨, 朱 玮, 魏 琪, 林志奎. 物理化学学报, 2015, 31, 1815.] doi: 10.3866/PKU.WHXB20150720
(45) Erlebacher, J. J. Electrochem. Soc. 2004, 151 (10), C614.doi: 10.1149/1.1784820
(46) Zhang, Z. H.; Wang, Y.; Qi, Z.; Zhang, W. H.; Qin, J. Y.; Frenzel, J.J. Phys. Chem. C 2009, 113 (29), 12629. doi: 10.1021/jp811445a
(47) Yeh, F. H.; Tai, C. C.; Huang, J. F.; Sun, I. W. J. Phys. Chem. B 2006,110 (11), 5215. doi: 10.1021/jp0552527
(48) Lei, W.; Briot, N. J.; Swartzentruber, P. D.; Balk, T. J. Metall. Mater.Trans. A 2014, 45 (1), 1. doi: 10.1007/s11661-013-2127-7
(49) Hayes, J. R.; Hodge, A. M.; Biener, J.; Hamza, A. V.; Sieradzki, K.J. Mater. Res. 2006, 21 (10), 2611. doi: 10.1557/jmr.2006.0322
(50) Renner, F. U.; Stierle, A.; Dosch, H.; Kolb, D. M.; Lee, T. L.;Zegenhagen, J. Nature 2006, 439 (7077), 707.doi: 10.1038/nature04465
(51) Chen, L. Y.; Chen, N.; Hou, Y.; Wang, Z. C.; Lv, S. H.; Fujita, T.; Jiang,J. H.; Hirata, A.; Chen, M. W. ACS Catal. 2013, 3 (6), 1220.doi: 10.1021/cs400135k
(52) Xu, C. X.; Su, J. X.; Xu, X. H.; Liu, P. P.; Zhao, H. J.; Tian, F.; Ding, Y.J. Am. Chem. Soc. 2007, 129 (1), 42. doi: 10.1021/ja0675503
(53) Liu, W.; Herrmann, A. K.; Bigall, N. C.; Rodriguez, P.; Wen, D.;Oezaslan, M.; Schmidt, T. J.; Gaponik, N.; Eychmuller, A. Acc. Chem.Res. 2015, 48 (2), 154. doi: 10.1021/ar500237c
(54) Geng, D. S.; Ding, N.; Hor, T. S. A.; Liu, Z. L.; Sun, X. L.; Zong, Y.J. Mater. Chem. A 2015, 3 (5), 1795. doi: 10.1039/c4ta06008c
(55) Qi, Z.; Weissmüller, J. ACS Nano 2013, 7 (7), 5948.doi: 10.1021/nn4021345
(56) Gan, L.; Heggen, M.; O'Malley, R.; Theobald, B.; Strasser, P. Nano Lett. 2013, 13 (3), 1131. doi: 10.1021/nl304488q
(57) Cheng, I. C.; Hodge, A. M. J. Porous Mater. 2014, 21 (4), 467.doi: 10.1007/s10934-014-9793-8
(58) Cahn, J. W. Acta Metall. 1961, 9 (9), 795.doi: 10.1002/9781118788295.ch11
(59) Park, M.; Harrison, C.; Chaikin, P. M.; Register, R. A.; Adamson, D. H.Science 1997, 276 (5317), 1401. doi: 10.1126/science.276.5317.1401
(60) Fukutani, K.; Tanji, K.; Motoi, T.; Den, T. Adv. Mater. 2004, 16 (16),1456. doi: 10.1002/adma.200400268
(61) Ding, Y.; Chen, M. W. MRS Bull. 2009, 34 (8), 569.doi: 10.1557/mrs2009.156
(62) Ding, Y.; Chen, M.; Erlebacher, J. J. Am. Chem. Soc. 2004, 126 (22),6876. doi: 10.1021/ja0320119
(63) Fujita, T.; Tokunaga, T.; Zhang, L.; Li, D.; Chen, L.; Arai, S.;Yamamoto, Y.; Hirata, A.; Tanaka, N.; Ding, Y. Nano Lett. 2014, 14 (3),1172. doi: 10.1021/nl403895s
(64) Fujita, T.; Okada, H.; Koyama, K.; Watanabe, K.; Maekawa, S.; Chen,M. W. Phys. Rev. Lett. 2008, 101 (16), 166601.doi: 10.1103/PhysRevLett.101.166601
(65) Xia, R.; Wang, J. L.; Wang, R. Y.; Li, X. D.; Zhang, X.; Feng, X. Q.;Ding, Y. Nanotechnology 2010, 21 (8), 085703.doi: 10.1088/0957-4484/21/8/085703
(66) Fujita, T.; Qian, L. H.; Inoke, K.; Erlebacher, J.; Chen, M. W. Appl.Phys. Lett. 2008, 92 (25), 251902. doi: 10.1063/1.2948902
(67) Liu, P.; Guan, P.; Hirata, A.; Zhang, L.; Chen, L.; Wen, Y.; Ding, Y.;Fujita, T.; Erlebacher, J.; Chen, M. Adv. Mater. 2016, 28 (9), 1753.doi: 10.1002/adma.201504032
(68) Rӧsner, H.; Parida, S.; Kramer, D.; Volkert, C. A.; Weissmüller, J. Adv.Eng. Mater. 2007, 9 (7), 535. doi: 10.1002/adem.200700063
(69) Pia, G.; Brun, M.; Aymerich, F.; Delogu, F. J. Mater. Sci. 2017, 52 (2),1106. doi: 10.1007/s10853-016-0407-5
(70) Zhang, X.; Ding, Y. Catal. Sci. Technol. 2013, 3 (11), 2862.doi: 10.1039/c3cy00241a
(71) Gu, X. H.; Xu, L. Q.; Tian, F.; Ding, Y. Nano Res. 2009, 2 (5), 386.doi: 10.1007/s12274-009-9038-3
(72) Yan, X. J.; Xiong, H. Y.; Bai, Q. G.; Frenzel, J.; Si, C. H.; Chen, X. T.;Eggeler, G.; Zhang, Z. H. RSC Adv. 2015, 5 (25), 19409.doi: 10.1039/c4ra17014h
(73) Ding, Y.; Kim, Y. J.; Erlebacher, J. Adv. Mater. 2004, 16 (21), 1897.doi: 10.1002/adma.200400792
(74) Norskov, J. K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jonsson, H. J. Phys. Chem. B 2004, 108 (46), 17886.doi: 10.1021/jp047349j
(75) Zhang, J. L.; Vukmirovic, M. B.; Sasaki, K.; Nilekar, A. U.;Mavrikakis, M.; Adzic, R. R. J. Am. Chem. Soc. 2005, 127 (36), 12480.doi: 10.1021/ja053695i
(76) Koh, S.; Strasser, P. J. Am. Chem. Soc. 2007, 129 (42), 12624.doi: 10.1021/ja0742784
(77) Srivastava, R.; Mani, P.; Hahn, N.; Strasser, P. Angew. Chem. Int. Ed.2007, 46 (47), 8988. doi: 10.1002/anie.200703331
(78) Wang, D.; Zhao, P.; Li, Y. Sci. Rep. 2011, 1 (7), 37.doi: 10.1038/srep00037
(79) Oezaslan, M.; Heggen, M.; Strasser, P. J. Am. Chem. Soc. 2012, 134(1), 514. doi: 10.1021/ja2088162
(80) Li, X.; Chen, Q.; Mccue, I.; Snyder, J.; Crozier, P.; Erlebacher, J.;Sieradzki, K. Nano Lett. 2014, 14 (5), 2569. doi: 10.1021/nl500377g
(81) Snyder, J.; McCue, I.; Livi, K.; Erlebacher, J. J. Am. Chem. Soc. 2012,134 (20), 8633. doi: 10.1021/ja3019498
(82) Baldizzone, C.; Gan, L.; Hodnik, N.; Keeley, G. P.; Kostka, A.;Heggen, M.; Strasser, P.; Mayrhofer, K. J. J. ACS Catal. 2015, 5 (9),5000. doi: 10.1021/acscatal.5b01151
(83) Snyder, J.; Fujita, T.; Chen, M. W.; Erlebacher, J. Nat. Mater. 2010,9 (11), 904. doi: 10.1038/nmat2878
(84) Benn, E.; Uvegi, H.; Erlebacher, J. J. Electrochem. Soc. 2015, 162(10), H759. doi: 10.1149/2.0161510jes
(85) Snyder, J.; Livi, K.; Erlebacher, J. Adv. Funct. Mater. 2013, 23 (44),5494. doi: 10.1002/adfm.201301144
(86) Wang, R. Y.; Xu, C. X.; Bi, X. X.; Ding, Y. Energy Environ. Sci.2012, 5 (1), 5281. doi: 10.1039/c1ee02243a
(87) Duan, H. M.; Xu, C. X. Electrochim. Acta 2015, 152, 417.doi: 10.1016/j.electacta.2014.11.160
(88) Duan, H. M.; Hao, Q.; Xu, C. X. J. Power Sources 2015, 280, 483.doi: 10.1016/j.jpowsour.2015.01.136
(89) Xu, C. X.; Zhang, H.; Hao, Q.; Duan, H. M. ChemPlusChem 2014,79 (1), 107. doi: 10.1002/cplu.201300311
(90) Han, B. H.; Xu, C. X. Int. J. Hydrogen Energy 2014, 39 (32), 18247.doi: 10.1016/j.ijhydene.2014.09.006
(91) Chen, X. T.; Jiang, Y. Y.; Sun, J. Z.; Jin, C. H.; Zhang, Z. H. J.Power Sources 2014, 267 (4), 212. doi:10.1016/j.jpowsour.2014.05.089
(92) Zhang, Z. H.; Wang, Y.; Wang, X. G. Nanoscale 2011, 3 (4), 1663.doi: 10.1039/c0nr00830c
(93) Zhang, Z. H.; Zhang, C.; Sun, J. Z.; Kou, T. Y.; Bai, Q. G.; Wang, Y.;Ding, Y. J. Mater. Chem. A 2013, 1 (11), 3620.doi: 10.1039/c3ta01464a
(94) Lang, X. Y.; Han, G. F.; Xiao, B. B.; Gu, L.; Yang, Z. Z.; Wen, Z.;Zhu, Y. F.; Zhao, M.; Li, J. C.; Jiang, Q. Adv. Funct. Mater. 2015, 25(2), 230. doi: 10.1002/adfm.201401868
(95) Zeis, R.; Lei, T.; Sieradzki, K.; Snyder, J.; Erlebacher, J. J. Catal.2008, 253 (1), 132. doi: 10.1016/j.jcat.2007.10.017
(96) Shao, M. J. Power Sources 2011, 196 (5), 2433.doi: 10.1016/j.jpowsour.2010.10.093
(97) Xu, C. X.; Zhang, Y.; Wang, L. Q.; Xu, L. Q.; Bian, X. F.; Ma, H. Y.;Ding, Y. Chem. Mater. 2009, 21 (14), 3110.doi: 10.1021/cm900244g
(98) Xu, C. X.; Wang, L. Q.; Wang, R. Y.; Wang, K.; Zhang, Y.; Tian, F.;Ding, Y. Adv. Mater. 2009, 21 (21), 2165.doi: 10.1002/adma.200702700
(99) Zhang, H.; Hao, Q.; Geng, H. R.; Xu, C. X. Int. J. Hydrogen Energy 2013, 38 (24), 10029. doi: 10.1016/j.ijhydene.2013.06.010
(100) Yang, R. Z.; Bian, W. Y.; Strasser, P.; Toney, M. F. J. Power Sources 2013, 222 (2), 169. doi: 10.1016/j.jpowsour.2012.08.064
(101) Xu, C.; Liu, Y.; Zhang, H.; Geng, H. Chem. - Asian J. 2013, 8 (11),2721. doi: 10.1002/asia.201300607
(102) Xu, C. X.; Liu, Y. Q.; Hao, Q.; Duan, H. M. J. Mater. Chem. A 2013,1 (43), 13542. doi: 10.1039/c3ta12765f
(103) Liu, Y.; Xu, C. ChemSusChem 2013, 6 (1), 78.doi: 10.1002/cssc.201200752
(104) Zhou, Y.; Lu, Q.; Zhuang, Z. B.; Hutchings, G. S.; Kattel, S.; Yan, Y.S.; Chen, J. G.; Xiao, J. Q.; Jiao, F. Adv. Eng. Mater. 2015, 5 (13),1500149. doi: 10.1002/aenm.201500149
(105) Xu, C. X.; Li, Y. Y.; Tian, F.; Ding, Y. ChemPhysChem 2010, 11(15), 3320. doi: 10.1002/cphc.201000313
(106) Gu, X.; Cong, X.; Ding, Y. ChemPhysChem 2010, 11 (4), 841.doi: 10.1002/cphc.200900927
(107) Chen, B.; Meng, G. W.; Huang, Q.; Huang, Z. L.; Xu, Q. L.; Zhu, C.H.; Qian, Y. W.; Ding, Y. ACS Appl Mater Inter 2014, 6 (18), 15667.doi: 10.1021/am505474n
Nanoporous Metal Electrocatalysts for Oxygen Reduction Reactions
ZHAI Xiao1,3DING Yi2,3,*
(1School of Materials Science and Engineering, University of Shanghai for Science and Technology, Shanghai 200093, P. R. China;2Institute for New Energy Materials & Low-Carbon Technologies, School of Materials Science and Engineering, Tianjin University of Technology, Tianjin 300384, P. R. China;3Tianjin Key Laboratory of Advanced Functional Porous Materials, Tianjin 300384, P. R. China)
Fuel cells allow the direct conversion of the chemical energy in chemical fuels to electricity,with particular advantages of being highly effective, environment-friendly, and portable. For those fuel cells using oxygen or air as the oxidant, the oxygen reduction reaction (ORR) occurring on the cathode remains the major obstacle for the commercialization of fuel cell technologies because of its slow kinetics, which in turn results in relatively low catalytic efficiency and high price due to excessive use of precious metals like Pt. In recent years, dealloyed nanoporous metals have garnered widespread attention in the field of electrocatalysis due to their unique structural properties, such as three-dimensionally interconnected pore/ligament structure, excellent conductivity, and structural flexibility. This review summarizes the recent advances in nanoporous metal catalysts for ORR, with an emphasis on their unique structural properties for the development of new-generation high-performance fuel cell catalysts.
Nanoporous metal; Dealloying; Fuel cell; Oxygen reduction reaction; Low Pt catalyst
February 20, 2017; Revised: April 3, 2017; Published online: April 17, 2017.
O643
10.3866/PKU.WHXB201704173 www.whxb.pku.edu.cn
*Corresponding author. Email: yding@tjut.edu.cn.
The project was supported by the National Natural Science Foundation of China (51671145).国家自然科学基金(51671145)资助项目
© Editorial office of Acta Physico-Chimica Sinica
翟萧,2013年本科毕业于上海理工大学材料科学与工程学院,2015年至今为上海理工大学材料科学与工程学院硕士研究生,并在天津市先进多孔功能材料重点实验室联合培养。主要研究方向为纳米多孔金属电催化剂的制备及应用。
丁轶,天津理工大学新能源材料与低碳技术研究院教授。主要从事纳米多孔金属材料的研发及在能源、资源、环境等领域中的应用基础研究。发表学术论文100余篇,被引用7300余次,个人H指数45,授权国内外专利30余项。
