Separation and purification of acteoside from Rehmanniaglutinosa by combining macroporous resin with high-speed countercurrent chromatography
2017-09-14BUZhisiHEQingZHAORushiCHUChuLIXingnuoTONGShengqiang
BU Zhisi, HE Qing, ZHAO Rushi, CHU Chu, LI Xingnuo, TONG Shengqiang
(College of Pharmaceutical Science, Zhejiang University of Technology, Hangzhou 310032, China)
Article
Separation and purification of acteoside fromRehmanniaglutinosaby combining macroporous resin with high-speed countercurrent chromatography
BU Zhisi, HE Qing, ZHAO Rushi, CHU Chu, LI Xingnuo, TONG Shengqiang*
(College of Pharmaceutical Science, Zhejiang University of Technology, Hangzhou 310032, China)
Acteoside, a chemical component of the traditional Chinese medicinal herbRehmanniaglutinosa, was separated from the plant by a combination of macroporous resin column chromatography and high-speed countercurrent chromatography. The static adsorption and desorption of acteoside in the crude extract were investigated using four kinds of macroporous resin, among which the D101 macroporous resin presented the best adsorption and desorption rates toward the compound. The crude extract was then gradient-eluted with increa-sing volume percentages of ethanol, where it was found that the highest content of acteoside was obtained when using 10% (v/v) ethanol eluent, in which the purity of acteoside was increased from 4.9% to 32.6%. Then, the partially purified crude extract (165 mg) was further purified by high-speed countercurrent chromatography using a two-phase solvent system consisting of ethyl acetate-n-butanol-water (1∶4∶5, v/v/v), yielding 45 mg of acteoside with 96% purity.
countercurrent chromatography; macroporous resin;Rehmanniaglutinosa; acteoside
Rehmanniaglutinosais a plant species in the family Scrophulariaceae, the fresh or dried roots of which have traditionally been used as a folk medicine in China. The plant is distributed in the Chinese provinces of Liaoning, Hebei, Henan, Shandong, Shanxi, Gansu and Neimenggu. Three preparations ofRehmanniaglutinosaare used as medicinalmaterials: fresh rehmannia root, dried rehmannia rhizome and prepared rehmannia root. The pharmacological activities ofRehmanniaglutinosainclude effects on the blood system, immune system, cardiovascular system, endocrine system, kidneys and nervous system, and reinforcement of learning and memory [1,2]. The plant contains a wide range of active chemical constituents, including iridoid glycosides, phenylethanoid glycosides, carbohydrates, amino acids and trace elements. The main phenylethanoid glycosides are acteoside, isoacteoside, cistanoside A, cistanoside F, purpureaside C, jionoside A1, jionoside B1 and echinacoside [3]. Phenylethanoid glycosides have been shown to present various bioactivities, including antitumor, antiviral, antibacterial and antioxidant effects, suppression of neuronal apoptosis, enhancement of memory [4]. In the newest edition of the pharmacopeia of traditional Chinese medicine [1], the concentrations of two components, acteoside and catalpol, are listed as the quality control criteria forRehmanniaglutinosa, indicating their importance as the main bioactive ingredients. Meanwhile, in view of their wide pharmacological activities, large quantities of pure compounds from this species are urgently needed as chemical refe-rence standards as well as for further pharmacological studies.

Fig. 1 Chemical structure of acteoside
In our previous work, the bioactive iridoid glycoside catalpol was successfully separated by high-speed countercurrent chromatography with high purity from the partially purified crude extract ofRehmanniaglutinosa[5]. Here, we report our subsequent separation of acteoside from this plant by a combination of macroporous resin and high-speed countercurrent chromatography. The chemical structure of acteoside is shown in Fig. 1. The traditional method of separating che-mical components from plants is silica gel column chromatography, which is generally time- and solvent-consuming and offers low separation efficiency. High-speed countercurrent chromatography is a support-free liquid-liquid partition chromatography that eliminates the shortcoming of irreversible adsorption in traditional methods, thereby reaching product recoveries of almost 100%. Furthermore, the cost of separation is ge-nerally lower than that of traditional methods, and the technique is easily scaled up [6]. Therefore, this technique has become widely used in the separation of components from natural products. In total, six papers have been published that describe the separation of acteoside from various plants by countercurrent chromatography [7-12]. High contents of acteoside were found in all the reported plants, which allowed relatively easy separation of acteoside from the crude extracts by countercurrent chromatography. However, two-step separations, in which countercurrent chromatography was used with two different biphasic solvent systems, were necessary in most of these studies [8-10]. In another study, the sample was subjected to AB-8 resin column chromatography prior to countercurrent chromatography, but low peak resolution was found in this case [12]. A different study [7] used glacial acetic acid for the biphasic solvent system, but this proved difficult to remove after separation. Last but not least, we found no paper available that describes the separation of acteoside from the traditional Chinese herbRehmanniaglutinosa.
In our preliminary experiments [5], it was found that the chemical composition of the crude extracts ofRehmanniaglutinosawas extremely complicated, and the very low content of acteoside made it impossible to separate this component with high purity in one step by countercurrent chromatography. The crude extract contained a large number of phenylethanoid glycosides and iridoid glycosides with very similar chemical structures. Therefore, a suitable method for enrichment of the acteoside component was necessary before conducting its separation by countercurrent chromatography. In the present research, we report a method for one-step separation of acteoside fromRehmanniaglutinosaby countercurrent chromatography with high peak resolution after the enrichment of the target component by macroporous resin. Macroporous resin is a polymeric adsorbent for column chromatography. It has several advantages, including low price and ease of recycling, compared with other column chromatographic materials. The separation and purification of some components with very low content from natural products remains challenging. Therefore, a method combining macroporous resin column chromatography and high-speed countercurrent chromatography represents a possible alternative for the separation of certain glycosides with very low content from plants.
1 Experimental section
1.1Apparatus
A type-J high-speed countercurrent chromatography apparatus was used (Model TBE-200V, Shanghai Tauto Biotechnique, Shanghai, China). It was equipped with three upright multilayer coil separation columns connected in series. Each column consisted of 1.6 mm i. d. polytetrafluoroethylene tubing with a total capacity of 190 mL. The countercurrent chromatography centrifuge was placed in a vessel to allow the column temperature to be maintained by a Model SDC-6 constant-temperature controller (Ningbo Scientz Biotechnology Co. Ltd., Ningbo, China). The solvents were pumped into the column with a model s-1007 constant-flow pump (Beijing Shengyitong Technique, Beijing, China). Continuous monitoring of the effluent was achieved with a model UVD-200 UV detector (Shanghai Jinda Biotechnology Co., Ltd., Shanghai, China), and an SEPU3010 workstation (Hangzhou Puhui Techno-logy, Hangzhou, China) was employed to record the chromatograms.
High-performance liquid chromatography (HPLC) was performed using a Waters Breeze system (Waters Corporation, Milford, USA) comprising a Waters2487 dualλabsorbance detector, a Waters717 plus autosampler, a Waters1525 controller, a Waters1525 binary HPLC pump and a Breeze workstation.
1.2Reagents and materials
Analytical grade ethanol was purchased from Hangzhou Huipu Chemical Instrument Co., Ltd., Hangzhou, China, and used for the extraction ofRehmanniaglutinosaand for the pretreatment of the macroporous resins. Methanol and acetonitrile of chromatographic grade used in the HPLC system were purchased from Tedia (Fairfield, USA). The macroporous resins AB-8, D101, HPD300 and ADS-17 were purchased from Cangzhou Baoen Chemical Co., Ltd., Cangzhou, China.Rehmanniaglutinosawas purchased from Jiuzhou Pharmacy, Hangzhou, China. A reference sample of acteoside was purchased from Chengdu Mann Stewart Biological Technology Co. Ltd., Chengdu, China.
1.3Extraction and separation methods
1.3.1Preparation of crude extract
The dried root ofRehmanniaglutinosawas ground into a fine powder, commonly known asRadixrehmanniae. Then, 300 g ofRadixrehmanniaewas placed in a 1 000 mL round flask and extracted with 500 mL of 70% (v/v) ethanol aqueous solution under reflux, twice (for 1.5 h each time). The mixed decoction was filtered and concentrated at 60 ℃ under reduced pressure, yielding about 40 g of the crude extract.
1.3.2Pretreatment of the macroporous resins
The wet macroporous resins were oven-dried at 60 ℃ for 24 h. Then, they were soaked in 95% (v/v) ethanol for 24 h to remove impurities and washed successively with 5% (v/v) HCl solution, deionized water and 5% (m/m) NaOH solution. Finally, they were washed with deionized water to remove the monomers and porogenic agents trapped inside the pores during the preparation process of the macroporous resins.
1.3.3Static adsorption and desorption
Macroporous resin column chromatography is an established technique for preferential enrichment of natural glycosides by elimination of highly polar water-soluble impurities [12-14]. Therefore, macroporous resin was pre-loaded into the column before purification of acteoside by countercurrent chromatography. For separation of the target compound by macroporous resin column chromatography, selection of a suitable macroporous resin was the first step. A good adsorbent should have a large adsorption capacity as well as high desorption ratio(s) toward the target compound(s). The static adsorption and desorption of acteoside in the crude extract using the four kinds of macroporous resin were investigated as follows. The resins (equal to 1.0 g of dry resins) were put into a flask, and 10.0 mL aliquots of sample solutions of the crude extract were added. These sample solutions were prepared by dissolving the crude extract in water, such that the initial concentrations of the target compound in the sample solutions were 40 g/L. The flasks were kept at room temperature for 24 h to reach adsorption equilibrium. The solutions were analyzed by HPLC after static adsorption. The adsorption capacities were calculated based on Equation (1) and the desorption capacities were calculated according to Equation (2). After reaching adsorption equilibrium, the resins were desorbed with 10.0 mL of 95% (v/v) ethanol. The flasks were kept at room temperature for 24 h. The desorption ratios were then calculated according to Equation (3). All adsorption and desorption experiments were carried out at least three times.
(1)
(2)
(3)
whereqeis the adsorption capacity (mg/g resin),C0andCeare the initial and equilibrium mass concentrations of acteoside (g/L),Viis the volume of the initial sample solution (mL),Wis the mass of the tested dry resin (g),qdis the desorption capacity (mg/g resin),Cdis the mass concentration of acteoside in the desorption solution (g/L),Vdis the volume of the desorption solution (mL) andDis the desorption ratio (%).
1.3.4Macroporous resin column chromatography
The resins after pretreatment (equal to 10.0 g dry resins) were loaded into a macroporous resin column. And 250 mL 184.9 g/L sample solutions were added. The sample solution was prepared by dissolving the crude extract in water. The flow rate was controlled at about 4 times the volume of the resin per hour. The column was first washed with water to remove the water-soluble components, and then eluted with 5%, 10%, 15%, 20%, 40%, 60%, 80% and 95% (v/v) ethanol, successively. Then, each collected fraction was analyzed by HPLC, and the fraction with the highest content of the target component was found to be that eluted with 10% (v/v) ethanol. Therefore, a further series of fractions was collected using 10% (v/v) ethanol eluent and evaporated to dryness under reduced pressure at 60 ℃. A partially purified crude sample was obtained, ready for further purification by high-speed countercurrent chromatography.
1.3.5Selection of two-phase solvent system for countercurrent chromatography
The two-phase solvent system was selected according to the partition coefficient (K) of the target compound. TheKvalues were determined by HPLC as follows. Two-phase solvent systems with various proportions of different solvents were prepared and equilibrated. After phase separation, a small amount of crude sample was added into 2 mL of the equilibrated aqueous phase, which was then analyzed by HPLC. Then, 2 mL of organic phase was added and mixed. The mixture was shaken violently to reach partition equilibrium. Subsequently, the chemical composition of the aqueous phase was analyzed again by HPLC and the chemical composition of the organic phase was calculated by the subtraction method. TheKvalue was defined as the peak area of the target compound in the upper phase divided by the peak area of the target compound in the lower phase.
1.3.6Preparation of two-phase solvent system and sample solution
The selected two-phase solvent system, composed of ethyl acetate-n-butanol-water (1∶4∶5, v/v/v), was thoroughly equilibrated in a separation funnel by repeated vigorous shaking at room temperature. The two phases were separated and subjected to ultrasonic degassing for 10 min before use. The sample solution for separation by countercurrent chromatography was prepared by dissolving 165 mg of the partially purified crude sample, which had been obtained by enrichment with the D101 macroporous resin, in 10 mL of the mixture solution of the lower phase and upper phase (1∶1, v/v) of the solvent system.
1.3.7Separation procedure of countercurrent chromatography
The multilayer coil column was entirely filled with the upper organic phase (stationary phase). Then, the lower aqueous phase (mobile phase) was pumped into the column at a flow rate of 1.5 mL/min. Then, the apparatus was rotated at 800 r/min. After hydrodynamic equilibrium was reached, as indicated by the emergence of the mobile phase, the sample solution was injected into the separation column through the injection valve. Data collection by the chromatography workstation began immediately. An automatic fraction collector was used to collect each eluted fraction, and each peak fraction was analyzed by HPLC. After separation, the fraction containing the target component was lyophilized to obtain the final product.
1.4Analytical method
The partially purified sample obtained from macroporous resin, and all peak fractions from countercurrent chromatography, were analyzed by HPLC. The analyses were performed with a Phenomenex GEMINI 5 μm C18110A column with 5 μm particle size of the packing material, 250 mm×4.6 mm i. d. (Phenomenex Company, Torrance, CA, USA). The mobile phases were composed of acetonitrile-water with gradient elution of acetonitrile (0-8 min, 12%-15%; 8-18 min, 15%-19%; 18-30 min, 19%-27%; 30-45 min, 27%-32%) at a flow rate of 0.6 mL/min, and the effluent was monitored by a UV detector at 334 nm. The column temperature was maintained at 25 ℃. Routine calculations of the amount of sample were made by comparison of the peak area with that of the standard.
1.5Identification of chemical structure
Identification of the peak fractions was carried out by1H NMR and comparison with the HPLC retention time of the standard compound. The1H NMR spectra were recorded on a Bruker AVANCE III 500MHz spectrometer (Bruker Corporation, Switzerland) with tetramethylsilane (TMS) as an internal standard.
2 Results and discussion
2.1Enrichment of acteoside by macroporous resin
In a previous study [12], AB-8 macroporous resin was used for the enrichment of acteoside; however, its adsorption and desorption capacity toward acteoside were not investigated, and no optimization of separation conditions was performed. In our study, four macroporous resins, including D101, AB-8, HPD300 and ADS-17, were investigated. The adsorption and desorption ratios of the target compound are shown in Fig. 2. From these results, it can be seen that D101, AB-8 and HPD300 all exhibited a much higher adsorption capacity toward acteoside than that of ADS-17, while D101 and ADS-17 showed the highest desorption capacities. Hence, D101 was the most suitable resin for the enrichment of acteoside from crude extract. As shown in Fig. 2, all four of the resins showed much lower desorption ratios toward the target component than their adsorption ratios. These high adsorption affinities may have been linked to the chemical structure of acteoside, in which four hydroxyl groups are attached to aromatic rings, enabling strong hydrogen bonding interactions with the resin during adsorption.

Fig. 2 Static adsorption and desorption capacity of the four macroporous resins toward the target component acteoside
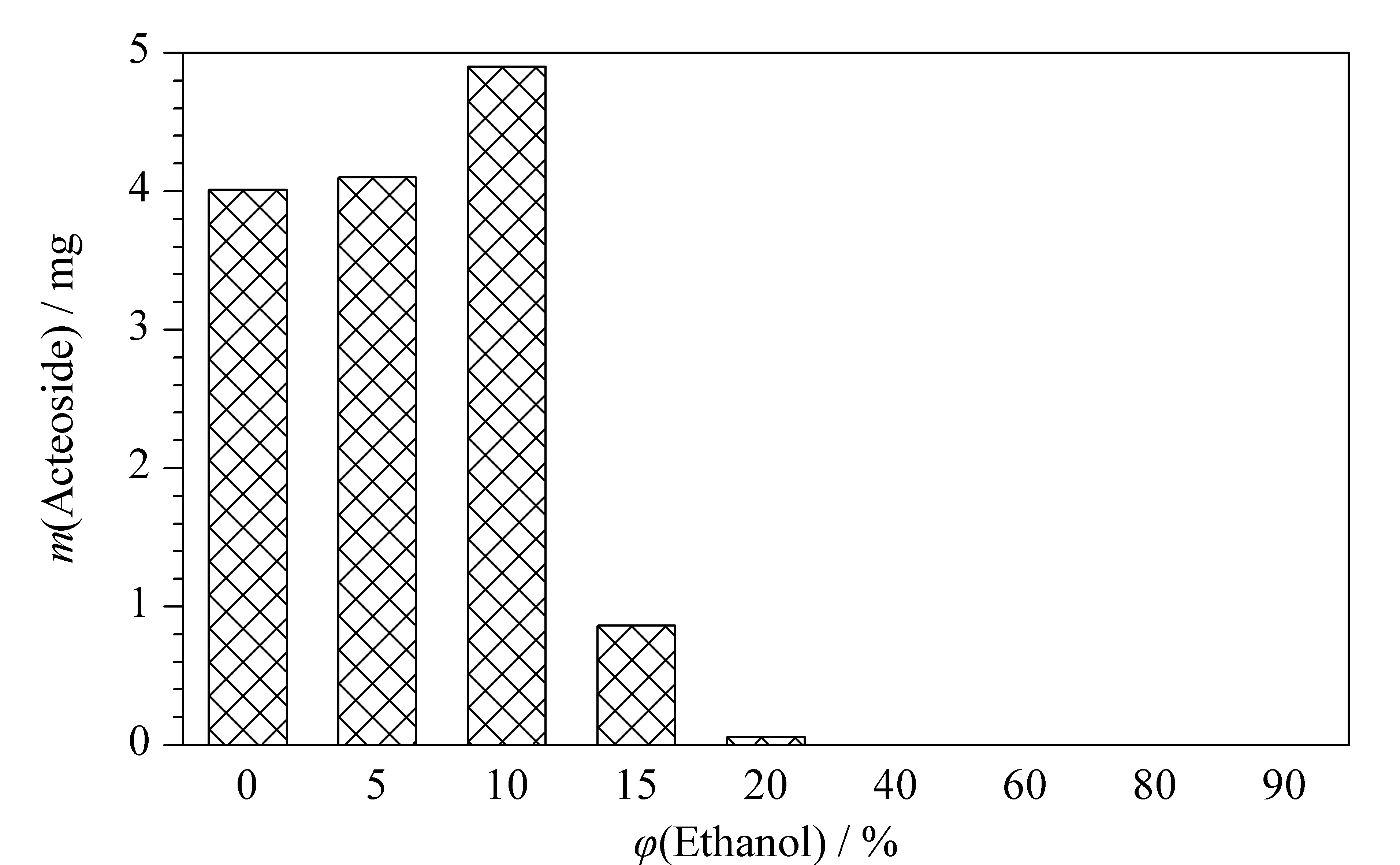
Fig. 3 Elution performance of D101 macroporous resin with regard to the content of acteoside in different volume percentages of ethanol
Gradient elution of acteoside with increasing ethanol concentrations was carried out by column chromatography with the D101 macroporous resin. The content of acteoside in each eluted fraction was analyzed by HPLC, as shown in Fig. 3. It can be seen that the target component was eluted predominantly in the 0-10% (v/v) ethanol fractions, which indicated that acteoside was highly polar. The fraction eluted with pure water was mainly composed of saccharide derivatives. The highest content of acteoside was found in the fraction eluted with 10% (v/v) ethanol, which contained 32.6% acteoside (compared with 4.9% in the crude extract) as determined by HPLC. The raw HPLC data are shown in Fig. 4. Fig. 4a confirmed that the content of acteoside in the crude extract was very low. The main components of this extract may have been saccharide derivatives eluted in the first 5 min of the HPLC process, since a reverse-phase stationary phase was used. However, the content of acteoside was greatly increased after its enrichment by the macroporous resin, as shown in Fig. 4b.
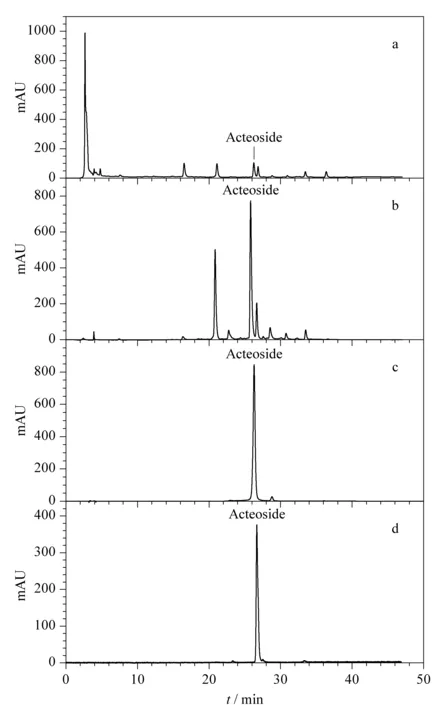
Fig. 4 Chromatograms of HPLC-UV analysis of (a) the ethanol crude extract of Rehmannia glutinosa, (b) the partially purified crude sample collected from 10% (v/v) ethanol elution by macroporous resin column chromatography, (c) the fraction containing the target component collected from countercurrent chromatography and (d) the acteoside reference sample Column: Phenomenex GEMINI 5 μm C18 110A column (5 μm particle size of packing material, 250 mm×4.6 mm i. d.); mobile phases: acetonitrile-water with gradient elution of acetonitrile (0-8 min, 12%-15%; 8-18 min, 15%-19%; 18-30 min, 19%-27%; 30-45 min, 27%-32%); column temperature: 25 ℃; flow rate: 0.6 mL/min; UV detector: 334 nm.
2.2Purification of acteoside by high-speed countercurrent chromatography
Successful separation of chemical components from natural products by high-speed countercurrent chromatography greatly depends on the selection of a suitable two-phase solvent system, since countercurrent chromatography works on the basis of liquid-liquid partition. Partition coefficients within the range 0.5-1.0 are generally considered acceptable for separation by high-speed countercurrent chromatography [5]. Large partition coefficients of the analytes generally lead to long retention times, resulting in peak broadening and a prolonged separation process, while small partition coefficients can lead to very short retention times, resulting in poor peak resolution. However, this is not always the case. For some purposes, the optimal solvent system may provide a partition coefficient far beyond the range normally considered acceptable. This is mainly dependent on the chemical composition of the crude sample: if the sample contains impurities with similar partition coefficients to the target analyte, this must be taken into consideration during the selection of the biphasic solvent system. Generally, in such cases, a large partition coefficient of the target component is preferable in order to obtain high peak resolution, despite the long retention time that results. Therefore, in order to improve the peak resolution of the target component in this study, a system was chosen in which the partition coefficient of the target component was sufficiently high to achieve complete separation. This was achieved with the aid of the exact peak-resolution equation for countercurrent chromatography, which was derived by Conway and Ito [15]:
(4)
whereαis the separation factor,Nis the number of theoretical plates for high-speed countercurrent chromatography,K1is the partition coefficient of the analytes andSFis the retention of the stationary phase. Equation (4) can be used to predict the peak resolution,Rs.Ncan be directly estimated from the chromatogram using the following equation:
(5)
whereVRis the retention volume of the analytes,Wbis the peak width. For the same two-phase solvent system and the same chromatographic apparatus,α,NandSFare roughly constant. Therefore, the value ofK1plays the major role in determining the peak resolution. The larger the partition coefficientK1, the better the peak resolution that can be obtained.
For the present separation, several typical biphasic solvent systems used for separation of compounds with large polarity were available, includingn-butanol-water (1∶1, v/v), ethyl acetate-n-butanol-water with different volume ratios,n-hexane-n-butanol-water with different volume ratios and aqueous-aqueous solvent systems. The aqueous-aqueous solvent system was not investigated in our study because of its high viscosity and difficulty of removal. The following solvent systems with different volume ratios were investigated with respect to the partition coefficient of acteoside:n-butanol-water (1∶1, v/v), ethyl acetate-n-butanol-water (4∶1∶5; 3∶2∶5; 2∶3∶5 and 1∶4∶5, v/v/v) andn-hexane-n-butanol-water (1∶4∶5, v/v/v). Each of the above solvent systems was tested on the same countercurrent chromatography apparatus. The results showed that a high peak resolution along with a suitable retention time of acteoside was obtained using the solvent system ethyl acetate-n-butanol-water (1∶4∶5, v/v/v), in which the partition coefficient of acteoside was found to be around 3.10, as determined by liquid-liquid partition experiments (see section 1.3.5). Fig. 5 shows a typical chromatogram for the separation of acteoside from the partially purified crude extract using the TBE-200V countercurrent chromatography apparatus. Approximately 165 mg of the partially purified crude sample collected from the D101 macroporous resin was dissolved in 10 mL of the mobile phase of the solvent system, yielding 45 mg of acteoside with over 96% purity after the separation, as indicated in Fig. 4c. The retention of the stationary phase was 55%, and the total separation time was 500 min.
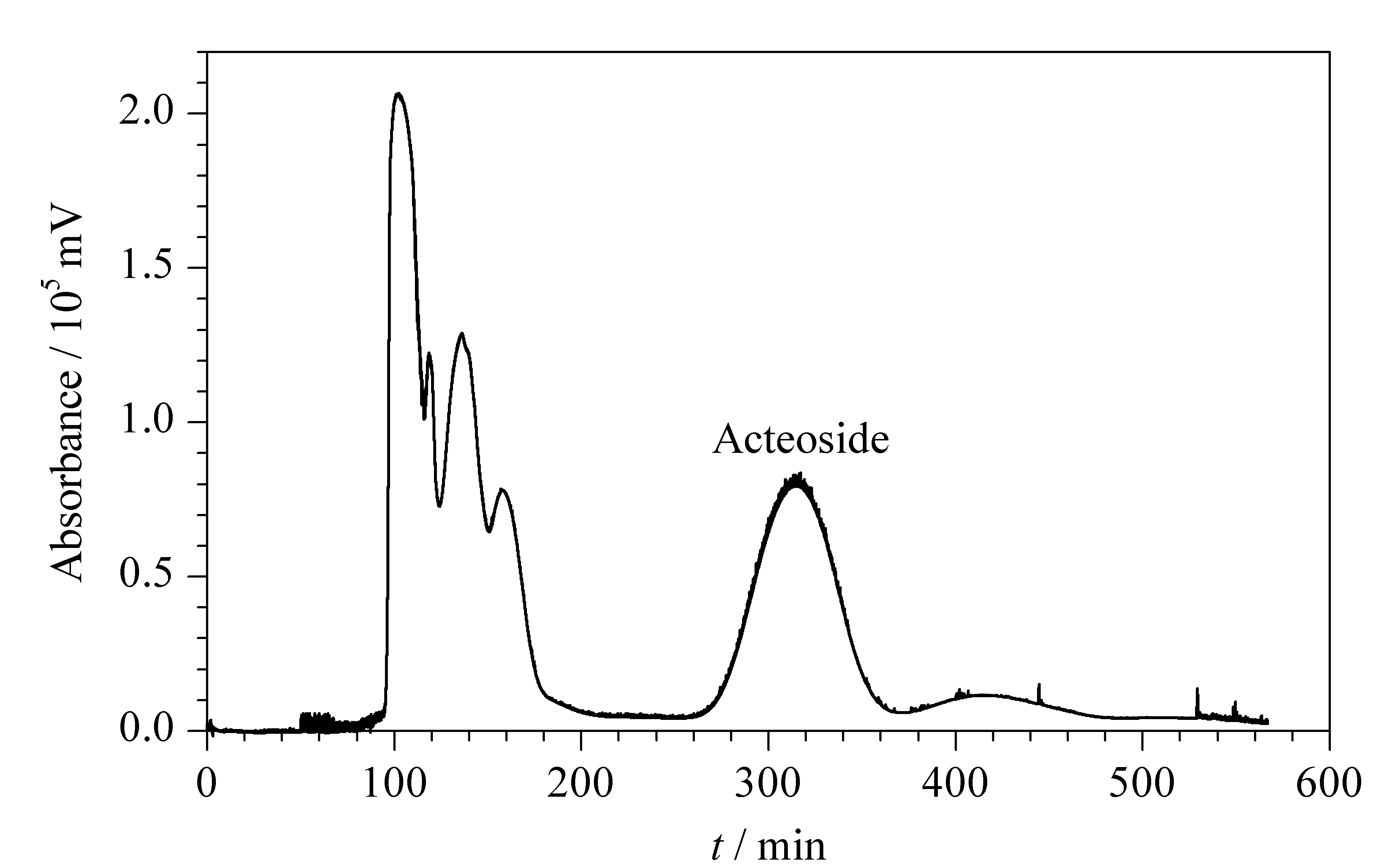
Fig. 5 Chromatogram for the separation of acteoside from the partially purified crude sample of Rehmannia glutinosa by high-speed countercurrent chromatography Solvent system: ethyl acetate-n-butanol-water (1∶4∶5, v/v/v); stationary phase: upper organic phase; mobile phase: lower aqueous phase; flow rate: 1.5 mL/min; revolution speed: 800 r/min; sample: 165 mg dissolved in 10 mL mixture solution of lower phase and upper phase (1∶1, v/v/v) of the solvent system; retention of the stationary phase: 55%.
According to the published literature [12], major differences in chemical composition between different species of traditional Chinese herbs can be found, including large differences in the content of acteoside. In an earlier study, the content of acteoside was found to be more than 40% in the crude extract from the plantPenstemonbarbatus. However, a much lower content of acteoside was found inRehmanniaglutinosain the present study, i. e., 4.9% (Fig. 4a), which indicated the greater difficulty of purifying acteoside from the crude extract of this plant. However, higher peak resolution for the target component was achieved in our work than that obtained previously. No baseline separation of acteoside was obtained in the earlier study, which led to low purity of the target component [12]. However, complete baseline separation of acteoside from the partially purified crude extract was obtained here by high-speed countercurrent chromatography, as shown in Fig. 5.
2.3Structural identification of acteoside
The structural identification of acteoside was carried out by1H NMR analysis and comparison with the HPLC retention time of the standard compound. The1H NMR spectral data of purified acteoside were as follows, acteoside: light yellow crystal, molecular formula C29H36O15.1H NMR (CD3OD/TMS, 500 MHz)δ: 7.61 (1H, d,J=15.9 Hz, H-7), 7.07 (1H, d,J=2.0 Hz, H-2), 6.97 (1H, dd,J=8.2, 2.0 Hz, H-6), 6.79 (1H, d,J=8.2 Hz, H-5), 6.29 (1H, d,J=15.9 Hz, H-8) for caffeoy; 6.71 (1H, d,J=2.0 Hz, H-2), 6.69 (1H, d,J=8.0 Hz, H-5), 6.58 (1H, dd,J=8.0, 2.0 Hz, H-6), 4.07 (1H, m, H-8), 3.93 (1H, m, H-8), 3.74 (1H, m, H-7), 3.64 (1H, m, H-7) for aglycone; 5.20 (1H, d,J=1.6 Hz, Rha-H-1), 4.40 (1H, d,J=7.9 Hz, Glu-H-1), 1.11 (3H, d,J=6.2 Hz, Rha-H-6), 2.82-4.95 (m, sugar-H) for sugar moiety.
3 Conclusions
A convenient and efficient method was established for the separation and purification of acteoside fromRehmanniaglutinosausing D101 macroporous resin combined with high-speed countercurrent chromatography. Acteoside has traditionally been separated from the plant by repeated silica gel column chromatography, which is time- and solvent-consuming. Moreover, separation of acteoside with high purity from the crude extract has proven impossible by one-step high-speed countercurrent chromatography because of the coexistence of many chemical constituents with very similar chemical structures and the very low content of acteoside in this plant. Herein, the combination of macroporous resin and high-speed countercurrent chromatography was proved to be an efficient alternative strategy for separation of this natural glycoside.
[1] Pharmacopoeia Commission of the People’s Republic of China. Pharmacopoeia of the People’s Republic of China, Part 1.10th ed. Beijing: The Medicine Science and Technology Press of China, 2015: 125
[2] Zhang R X, Li M X, Jia Z P. J Ethnopharmacol, 2008, 117: 199
[3] Chen C H, Song T Y, Liang Y C, et al. J Agric Food Chem, 2009, 57: 8852
[4] Yan F, Zeng G Y, Tian J B, et al. Central South Pharmacy, 2013, 11: 358
[5] Tong S Q, Chen L, Zhang Q, et al. J Chromatogr Sci, 2015, 53: 725
[6] Ito Y. J Chromatogr A, 2005, 1065: 145
[7] Xie C Y, Xu X J, Liu Q D, et al. J Liq Chromatogr Rel Technol, 2012, 35: 2602
[8] Lei L, Yang F Q, Zhang T Y, et al. J Liq Chromatogr Rel Technol, 2001, 24: 2187
[9] Li L, Tsao R, Liu Z Q, et al. J Chromatogr A, 2005, 1063: 161
[10] Li L, Tsao R, Yang R, et al. Food Chem, 2008, 108: 702
[11] Han L F, Ji L N, Boakye Y, et al. Molecules, 2012, 17: 8276
[12] Xie J, Tan F, Zhu J, et al. Food Chem, 2012, 135: 2536
[13] Geng S, Wang J Q, Xi X J, et al. Chinese Journal of Chromatography, 2017, 35(3): 302
[14] Zhao X H, Zhang Q X, Yuan X W, et al. Chinese Traditio-nal and Herbal Drugs, 2012, 43(10): 1971
[15] Conway W D, Ito Y. J Liq Chromatogr, 1985, 8(12): 2195
大孔树脂-高速逆流色谱法分离纯化地黄中毛蕊花糖苷
步知思, 何 青, 赵如诗, 楚 楚, 李行诺, 童胜强*
(浙江工业大学药学院, 浙江 杭州310032)
该文建立了大孔树脂-高速逆流色谱分离中药材地黄中有效成分毛蕊花糖苷的方法。考察了4种大孔树脂对地黄粗提物中毛蕊花糖苷的静态吸附与解吸情况,其中D101大孔树脂对目标成分的吸附率与解吸率最理想,实验结果表明体积分数为10%的乙醇洗脱得到的毛蕊花糖苷含量最高,目标成分含量从4.9%提高到32.6%。最后,部分纯化的样品(165 mg)采用高速逆流色谱进一步纯化,两相溶剂系统由乙酸乙酯-正丁醇-水(1∶4∶5, v/v/v)组成,分离得到45 mg纯度为96%的毛蕊花糖苷。
逆流色谱;大孔树脂;地黄;毛蕊花糖苷
10.3724/SP.J.1123.2017.04030
O658 Document code: A Article IC:1000-8713(2017)09-1014-08
* Received date: 2017-04-24
* Corresponding author. Tel/Fax: +86 571 88320984, E-mail: sqtong@zjut.edu.cn.
