中国尾凤蝶属昆虫(鳞翅目:凤蝶科)系统发育1)
2017-08-07易传辉赵健胡劭骥和菊冯志伟杨建华陈鹏
易传辉 赵健 胡劭骥 和菊 冯志伟 杨建华 陈鹏
(云南省林业科学院,昆明,650201) (西南林业大学) (云南大学) (云南省林业科学院)
中国尾凤蝶属昆虫(鳞翅目:凤蝶科)系统发育1)
易传辉 赵健 胡劭骥 和菊 冯志伟 杨建华 陈鹏
(云南省林业科学院,昆明,650201) (西南林业大学) (云南大学) (云南省林业科学院)
为探讨中国尾凤蝶属(Bhutanitis)昆虫的系统发生关系,采用线粒体DNACOI和ND1分子标记,对基因序列进行了分析,基于分子特征,采用NJ和贝叶斯法构建系统发育树。对3种尾凤蝶658 bp的COI基因序列的分析表明,三尾凤蝶(Bh.thaidina)、二尾凤蝶(Bh.mansfieldi)和多尾凤蝶(Bh.lidderdalii)T、C、A、G 4种核苷酸的平均含量分别为41.5%、40.0%、41.9%,15.2%、15.8%、14.3%,29.3%、30.7%、29.8%,14.0%、13.5%、14.0%;共发现突变位点131个,约占全长的19.91%,简约信息位点56个,约占全长的8.51%。对480 bp的NDI基因序列的分析表明,三尾凤蝶、二尾凤蝶和多尾凤蝶的NDI基因序列T、C、A、G的平均含量依次为34.4%、33.3%、32.1%,11.3%、11.9%、12.1%,45.6%、46.7%、47.1%,8.7%、8.1%、8.8%;共有63个变异位点,占全长的13.13%,简约信息位点39个,约占全长的8.13%。二者均表现为明显的A+T碱基偏向。中国分布尾凤蝶昆虫遗传多样性较低,种间遗传距离较小,系统发育分析表明,二尾凤蝶最早从祖先种群中分化,其次是多尾凤蝶,最后是三尾凤蝶;多尾凤蝶与其他二种亲缘关系均较近。
尾凤蝶;线粒体DNACOI基因;线粒体DNAND1基因;系统发育;遗传多样性
Bhutanitis; MitochondrialCOIgene; MitochondrialND1 gene; Phylogeny; Genetic diversity
尾凤蝶属(Bhutanitis)属鳞翅目(Lepidoptera)凤蝶科(Papilionidae)锯凤蝶亚科(Zerynthiinae)昆虫,为典型高山种类。分布于中国、印度、不丹、锡金、缅甸、泰国、越南等地,中国主要分布于云南、四川、陕西、甘肃和西藏等省(区)。目前,对该属物种存在争议,以周尧等[1]185-188,[2]8为代表的中国科学家认为该属有 7 种 2 亚种,即不丹尾凤蝶(Bh.ludlowi)、多尾凤蝶(Bh.lidderdalii)、三尾凤蝶(Bh.thaidina)(包括指名亚种(Bh.thaidinathaidina)和东川亚种(Bh.thaidinadongchuanensis)、玉龙尾凤蝶(Bh.Yulongensis)、玄裳尾凤蝶(Bh.nigrilima)、二尾凤蝶(Bh.mansfieldi)和丽斑尾凤蝶(Bh.pulchristriata)。以Hauser et al.[3]为代表的学者认为,尾凤蝶属应分为 4种,即不丹尾凤蝶、多尾凤蝶、三尾凤蝶和二尾凤蝶。除不丹尾凤蝶外,其余各种(亚种)均在中国有分布。目前该属昆虫已为濒危物种,为《濒危动植物进出口贸易公约》(CITES)限制贸易物种,同时被 IUGN 濒危物种红皮书和中国野生动物法列为Ⅱ级保护物种[1]185,[2]8,[3-4]。由于DNA的独特性,目前已成为物种区分的重要手段,线粒体基因因其不含间隔区和内含子,无重复序列,很少发生不等交换,在遗传过程中不易发生基因重组、倒位、易位等突变,已广泛应用于系统发育分析等研究[5-10]。诸立新等[11]利用COI基因部分序列对尾凤蝶属4种蝴蝶分子系统关系进行了探讨,结果表明,二尾种组与多尾凤蝶亲缘关系较近,而三尾凤蝶较早分化出来,并认为二尾和丽斑尾凤蝶可能为同一物种。笔者基于COI基因和ND1基因标记,采用MEGA5.0和Mrbayes 3.1.2软件,构建NJ树与贝叶斯树,对中国分布的尾凤蝶属3个物种系统发育进行分析,为深入研究该属昆虫的分子进化和保护提供基础资料。
1 材料与方法
1.1 供试昆虫
尾凤蝶属昆虫分别采自云南、四川和陕西3省,共49头;中华虎凤蝶(Luehdorfiachinensis)2头,来自浙江天目山。样本采集后,立即将一侧的中后足取下,放入无水乙醇的离心管中,并标记,野外采集回来后,置于冰箱中-20 ℃保存备用,标本信息见表1。
1.2 基因提取
将昆虫足移入1.5 mL的离心管中,加入1 mL STE缓冲液,在干式恒温器中37 ℃加热1 h,去除离心管中的缓冲液,重新加入200 μL STE缓冲液;用小剪刀将足剪碎,加入4 mL蛋白酶K;干式恒温器中37 ℃处理5 min,95 ℃加热15 min,每隔5 min震荡一次,待标本温度回到室温后4 000 r·min-1离心,冰箱中-40 ℃保存待用。
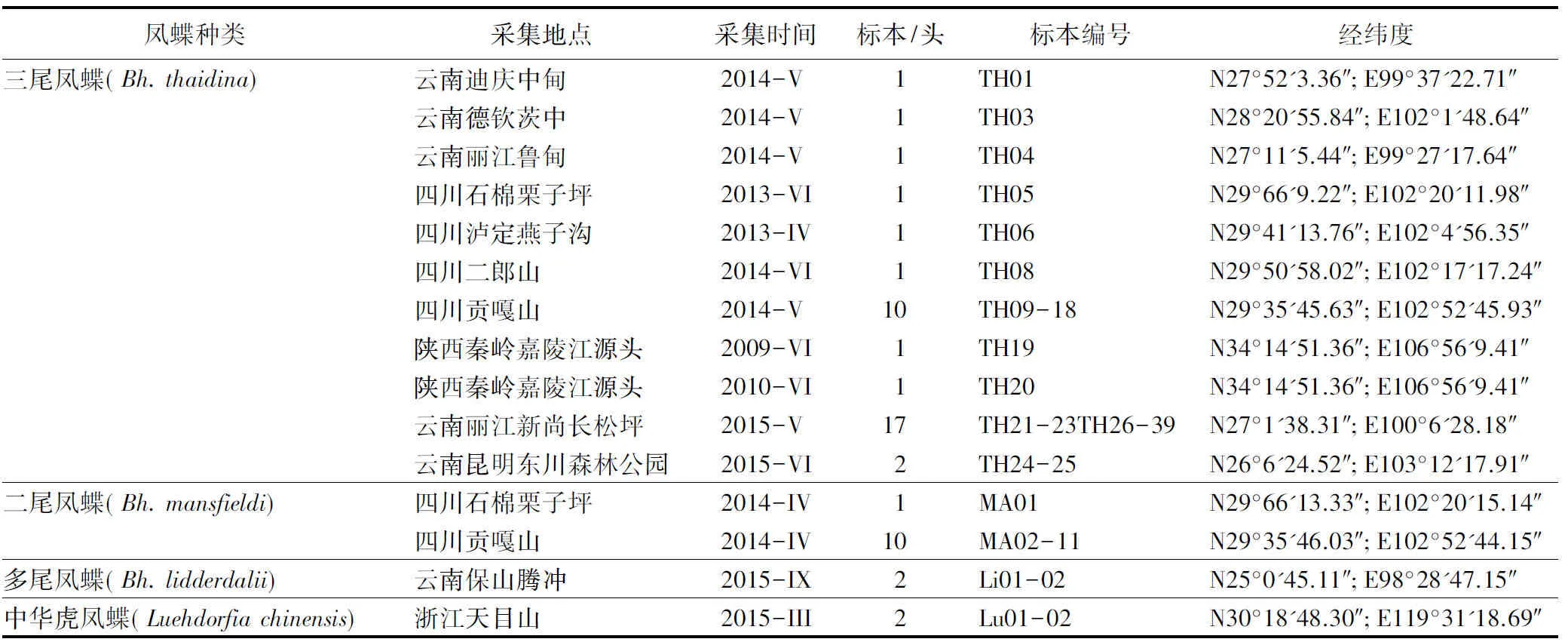
表1 尾凤蝶属昆虫标本采集信息
1.3 PCR反应与测序
扩增参照文献[12-15]的方法。COI引物序列:(Jerry)COI-F,5′-CAACATTTATTTTGATTTTTTGG-3′;(K698)CO1-F,5′-TACAATTT ATCGCCTAAACTTC AGCC-3′;(Pat2)COI-R,5′-TCCATTACATATAATCTGCCATATT-3′;(Djernaes)CO1-R,5′-GC TATTATAGCATA AATTATTCC-3′。NDI引物序列:NDI-F,5′-ATCAAAAGGAGCTCGATTAGTTTC-3′;NDI-R,5′-CGTAAAGTCCTAGGTTAT AT TCAGATTCG-3′。反应体系为25.00 μL,模板DNA1.00 μL,上下游引物各0.50 μL,ExTaqDNA聚合酶0.25 μL,dNTP Mix 和25.00 mmol MgCl2各2.00 μL,10×Loading Buffer 2.50 μL,加灭菌ddH2O 16.25 μL。扩增条件,预变性95 ℃ 3 min,变性94 ℃ 1 min,退火50 ℃ 1 min,延伸72 ℃ 1 min,30个循环,终延伸72 ℃ 5 min。反应结束后,取扩增产物2.4 μL,在1.5%琼脂糖凝胶上进行电泳检测(电压150 V,电泳缓冲液为0.5×TBE),在凝胶成像系统下观察并拍照记录。确定成功扩增出目标片断后,送上海赛音生物技术有限公司完成双向测序工作。COI与NDI使用的反应体系一致。
1.4 数据分析
从NCBI网站中通过BLAST同源检索确认所测标本的COI和NDI基因序列,并从NCBI数据库中下载目标序列,用于系统发生关系分析。校对后的测序结果通过BioEdit 7.0.9软件中的Clustal W功能进行逆向互补校对,并对序列进行手工矫正和剪切;利用MEGA 5.0对整理好的序列进行概念翻译,基于Kimura-2参数对序列特征、群体间遗传距离(P-distance)和遗传差异(Fst)进行分析[12-14]。应用DnaSP5.0软件分析计算三尾凤蝶各地理种群间单倍型多样性(Hd)、核苷酸平均差异数(k)和核苷酸多样性(Pi)[16]。
采用2种方法构建系统发育树[17-18],以中华虎凤蝶为外群,利用MEGA5.0软件基于Kimura-2-Parameter模型,用邻接法构建NJ系统发育树。用MrBayes 3.1.2软件建立BI系统发育树。最后利用软件SequenceMatrix-Windows-1.7.8对序列COI和NDI进行整合,并利用整合后的序列构建NJ树和BI树。
2 结果与分析
2.1 尾凤蝶属COI基因的特征与差异
三尾凤蝶各地理种群的两段COI部分序列,长分别为658、775 bp,二尾凤蝶和多尾凤蝶得到658 bp长度序列。对3种尾凤蝶658 bp序列进行分析表明,三尾凤蝶、二尾凤蝶和多尾凤蝶T、C、A、G核苷酸的平均含量依次为41.5%、15.2%、29.3%,14.0%、40.0%、15.8%,30.7%、13.5%、41.9%,14.3%、29.8%、14.0%。同源性比较显示,3种尾凤蝶中,共发现突变位点131个,约占全长的19.91%,简约信息位点56个,约占全长的8.51%;核苷酸有26个位点发生转换,占总替换数的68.42%,12个位点发生颠换,占31.58%,其中C与T之间的转换占总转换数的40.54%,总体转换/颠换偏倚率R值为2.706。尾凤蝶属种群间基于COI核苷酸多样性指数(Pi)为0.369 30,平均核苷酸差异数(Kxy)为243.000。
采用MEGA5.0软件,基于Kimura-2-Parameter双参数模型计算尾凤蝶和虎凤蝶各种群间的遗传距离。结果表明,尾凤蝶属种间的遗传距离(0.066~0.078)较小,与外群中华虎凤蝶之间的遗传距离(0.149~0.155)较大(表2)。
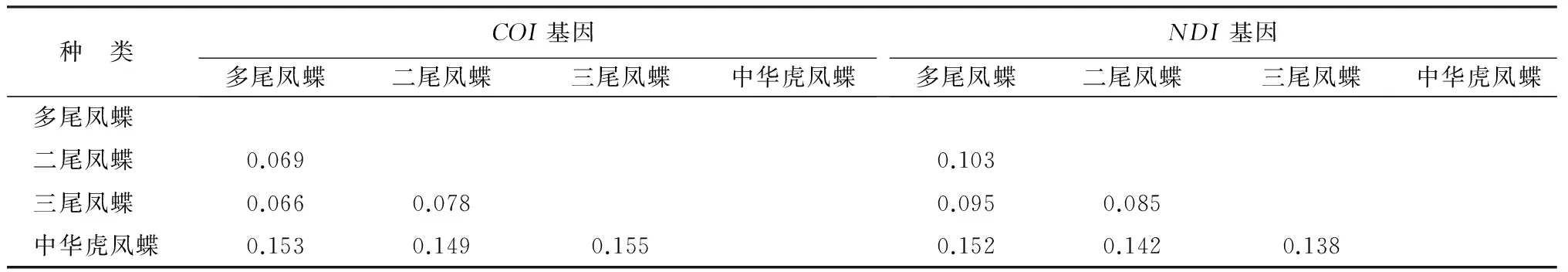
表2 尾凤蝶属各种间的遗传距离
2.2 尾凤蝶属NDI基因的特征与差异
通过整理去除测序结果两端侧翼序列,得线粒体NDI部分序列,长度为480 bp。三尾凤蝶、二尾凤蝶和多尾凤蝶该序列的T、C、A、G 4种核苷酸的平均含量依次为34.4%、11.3%、45.6%,8.7%、33.3%、11.9%,46.7%、8.1%、32.1%,12.1%、47.1%、8.8%。同源性比较显示,3种尾凤蝶中,共有63个变异位点,占全长的13.13%,保守位点417个,简约信息位点39个,约占全长的8.13%,自裔位点24个,未发现碱基缺失或插入。在所有核苷酸替换中,有14个位点发生转换,占总替换数的41.2%,发生颠换有20个位点,占58.8%,总体转换/颠换偏倚率R值为28.739。尾凤蝶属种群间基于NDI核苷酸多样性指数(Pi)为0.072 57,平均核苷酸差异数(Kxy)为34.833。
采用MEGA5.0软件,基于Kimura-2-Parameter双参数模型计算尾凤蝶和虎凤蝶各种群间的遗传距离。结果表明,尾凤蝶属种间遗传距离(0.085~0.103)略小于外群中华虎凤蝶种间遗传距离(0.138~0.152)(表2)。
2.3 系统发育
2.3.1 基于COI部分序列的分析
以线粒体COI基因部分序列为分子标记,以中华虎凤蝶为外群,采用MEGA5.0构建NJ树,采用Mrbayes 3.1.2构建贝叶斯树,对尾凤蝶属进行系统发育分析。2种方法得到的系统发育树极为相似,形成4个明显的分支,并具有较高的自展值。系统发育树表明,中华虎凤蝶与尾凤蝶属蝴蝶完全区分开,尾凤蝶属蝴蝶明显分为2个聚类簇,各个地理种群的三尾凤蝶个体首先聚在一起,随后与多尾凤蝶合成一个分支,二尾凤蝶的个体聚在一起形成另外一个分支。

分支下的数字是基于贝叶斯分析的后验率。
2.3.2 基于NDI部分序列的分析
基于所测尾凤蝶属线粒体NDI序列部分片段构建NJ树和贝叶斯树。2种方法得到的系统发育树极为相似,明显形成4个分支,并具有较高的自展值。系统发育树表明,中华虎凤蝶与尾凤蝶属蝴蝶明显分开,尾凤蝶属蝴蝶分为2个聚类簇,三尾凤蝶各地理种群首先聚在一起,然后和多尾凤蝶形成一个分支,二尾凤蝶的个体聚在一起形成另外一个分支,与基于COI序列所构建的系统发育树一致。
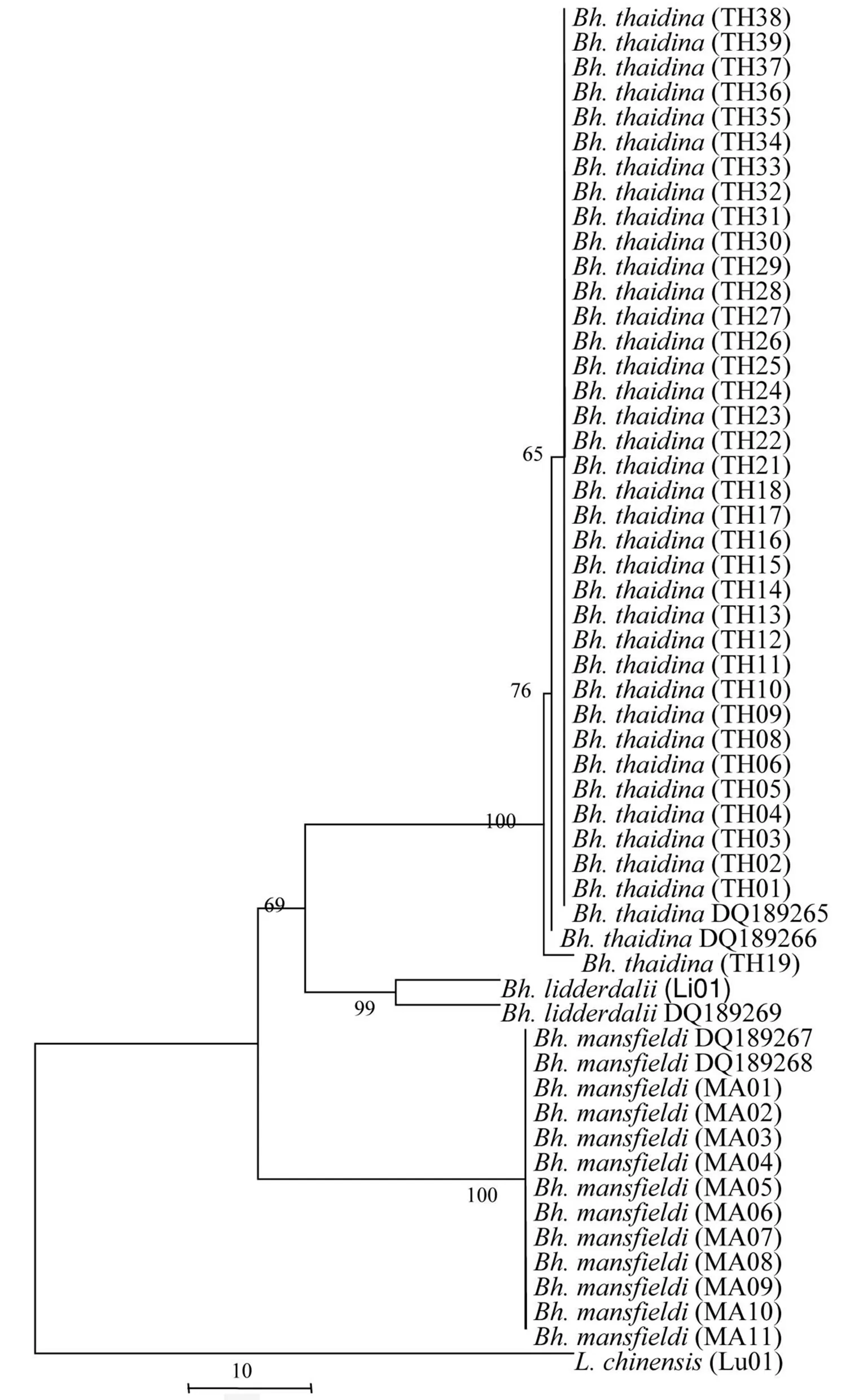
各分支上的数字为1 000次Bootstrap支持率;DQ为NCBI中检索到的尾凤蝶属COI序列片段。
图2 基于线粒体COI基因片段构建的NJ树

分支下数字是基于贝叶斯分析的后验率。
各分支上的数字为1 000次Bootstrap支持率。

分支下面的数字是基于贝叶斯分析的后验率。
图5 基于线粒体COI和NDI基因整合序列片段构建的贝叶斯树
2.3.3 拼接COI与NDI序列构建系统发育树
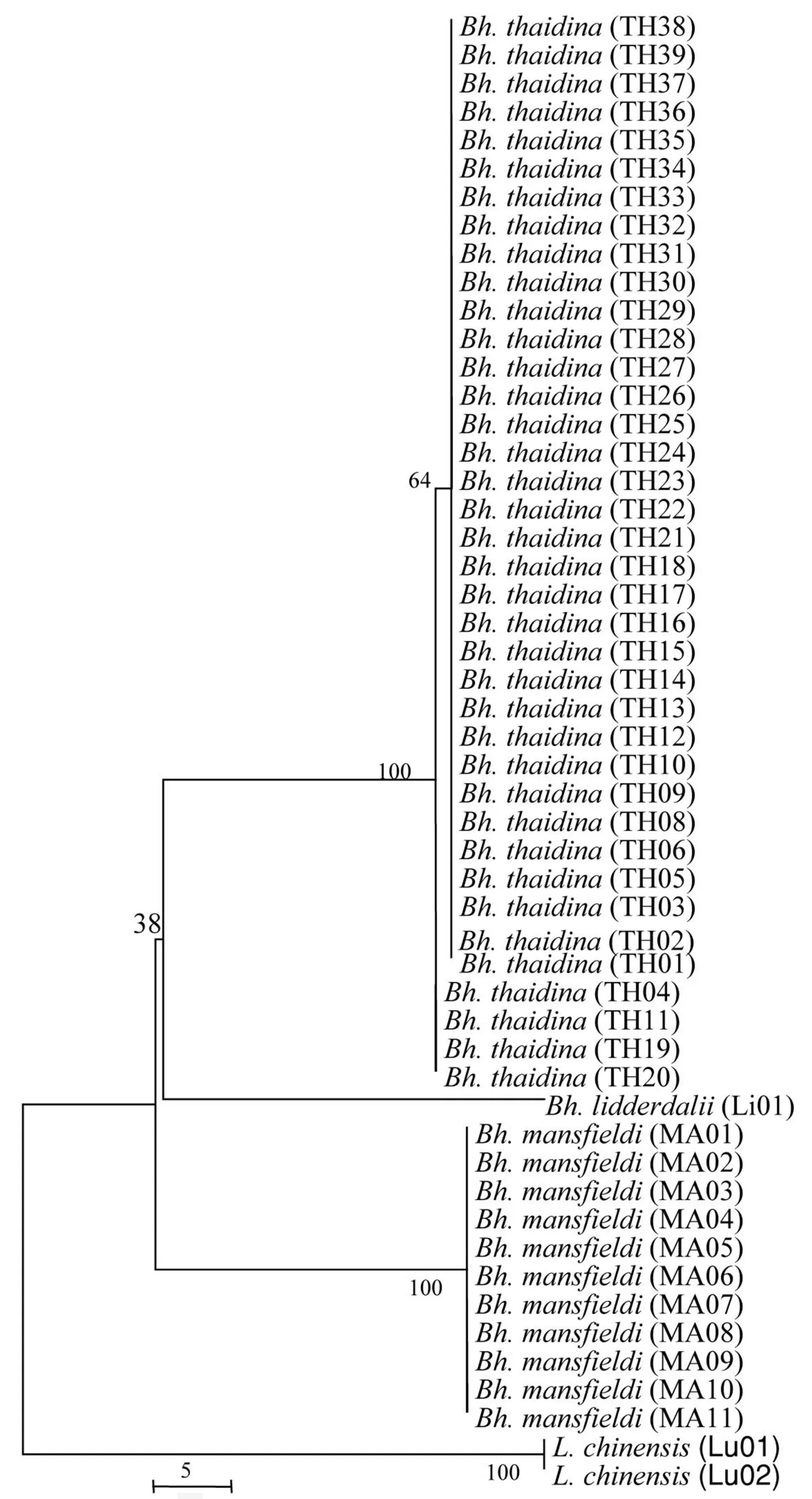
利用SequenceMatrix-Windows-1.7.8软件对各个样本的COI与NDI序列进行拼接,拼接后新序列总长度1 913 bp,构建贝叶斯树和NJ树。所得结论与COI和NDI序列分别单独构建的系统发育树几乎一致。
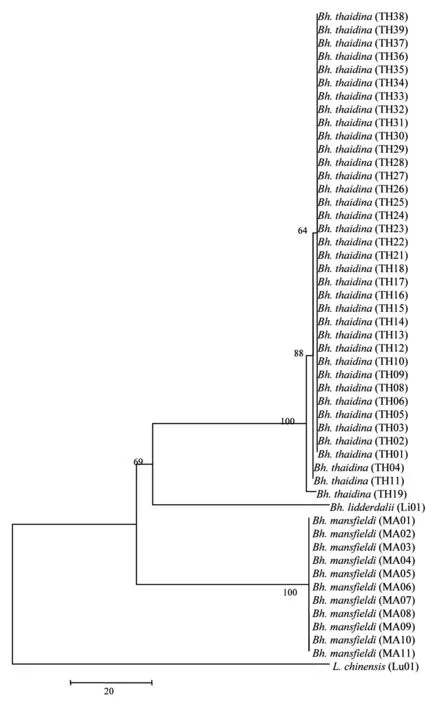
各分支上的数字为1 000次Bootstrap的支持率。
图6 基于线粒体COI和NDI基因整合序列片段构建的NJ树
3 结论与讨论
本研究利用线粒体COI和NDI基因序列,对中国分布的3种尾凤蝶属昆虫系统发育进行了研究,结果表明:(1)中国尾凤蝶属昆虫遗传多样性低,尾凤蝶属种群间基于COI核苷酸多样性指数(Pi)为0.369 30,基于NDI核苷酸多样性指数(Pi)为0.072 57,与诸立新等[12]的研究结论一致,表明该属昆虫适应性和生存能力较差,进化潜力弱。(2)二尾凤蝶最早从祖先种群中分化,其次是多尾凤蝶,最后是三尾凤蝶;二尾凤蝶与多尾凤蝶的亲缘关系相对较近,与三尾凤蝶的亲缘关系较远。结果与前人的研
究有一定差异,主要表现在多尾凤蝶在系统发育树中的位置,诸立新等的研究结果显示,多尾凤蝶最早从祖先中分化出来,其次是二尾凤蝶,最后是三尾凤蝶[12]。差异可能与研究标本数量、采集存放时间和保存方法有关。存放时间越长,对研究产生的影响越大。本研究利用线粒体COI和NDI基因序列进行分析得到的结果一致,可靠性较高,但由于部分种类个体数量较少,可能对研究结果有一定影响,有待今后补充并深入分析。
[1] 周尧.中国蝶类志[M].郑州:河南科学技术出版社,1999.
[2] 寿建新,周尧,李宇飞.世界蝴蝶分类名录[M].西安:陕西科学技术出版社,2006.
[3] 易传辉,和秋菊,王琳,等.三尾褐凤蝶的分布现状、濒危原因与保护性研究[J].湖北农业科学,2011,50(14):2851-2854.
[4] 李中文,符英丽.世界濒危及受保护的蝴蝶物种[J].海南师范学院学报(自然科学版),2000,13(2):102-107.
[5] 窦向梅,肖晖,黄大卫.DNA分类概述[J].生物学通报,2008,43(6):23-26.
[6] 杜启艳,常重杰.DNA条形码在鉴定物种中的应用[J].生物学教学,2010,35(12):60-61.
[7] 赵婉清,赵清,张虎芳.基于DNA条形码对菜蝽属内疑难种的鉴定(半翅目:蝽科)[J].山西农业大学学报(自然科学版),2015,35(2):169-174.
[8] 张民照,杜艳丽,张爱环,等.线粒体DNA标记在昆虫学研究中的应用进展[J].北京农学院学报,2011,26(1):76-80.
[9] 付景,张迎春.27种瓢虫mtDNA-COI基因序列分析及系统发育研究(鞘翅目:瓢虫科)[J].昆虫分类学报,2006,28(3):179-186.
[10] 代金霞,张大治.叶甲亚科部分种类COⅡ基因序列分析及系统发育初探[J].四川动物,2010,29(2):193-196.
[11] 诸立新,吴孝兵,晏鹏.基于COI基因部分序列对尾凤蝶属(鳞翅目,凤蝶科)四种蝴蝶分子系统关系及相关问题的探讨[J].动物分类学报,2006,31(1):25-30.
[12] AUBERT J, LEGAL L, DESCIMON H, et al. Molecular phylogeny of swallowtail butterflies of the tribe Papilionini (Papilionidae, Lepidoptera)[J]. Mol Phylogenet Evol,1999,12(2):156-167.
[13] THOMPSON J D, GIBSON T J, PLEWNIAK F, et al. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools[J]. Nucleic Acids Res,1997,25(24):4876-4882.
[14] TAMURA K, PETERSON D, PETERSON N, et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods[J]. Mol Biol Evol,2011,28(10):2731-2739.
[15] SIMONSEN TJ, ZAKHAROV EV, DJERNAES M, et al. Phylogenetics and divergence times of Papilioninae (Lepidoptera) with special reference to the enigmatic genera Teinopalpus and Meandrusa[J]. Cladistics,2010,27(2):113-137.
[16] SLATKIN M. A measure of population subdivision based on microsatellite allele frequencies[J]. Genetics,1995,139(1):457-462.
[17] RONQUIST F, HUELSENBECK J P. MrBayes 3: Bayesian phylogenetic inference under mixed models[J]. Bioinformatics,2003,19(12):1572-1574.
[18] DARRIBA D, TABOADA G L, DOALLO R, et al. jModelTest 2: more models, new heuristics and parallel computing[J]. Nat Methods,2012,9(8):772.
1)国家自然科学基金项目(31260527);云南省中青年学术技术带头人后备人才项目(2013HB093)。
易传辉,男,1970年8月生,云南省林业科学院,副研究员。E-mail:ynkcx2007@163.com。
2016年9月16日。
Q963;Q969.438.2
责任编辑:程 红。
Phylogenetic Analysis ofBhutanitis(Lepidoptera: Papiliondae) Distribution in China//Yi Chuanhui(Yunnan Academy of Forestry, Yunnan 650201, P. R. China); Zhao Jian(Southwest Forestry University); Hu Shaoji(Yunnan University); He Ju, Feng Zhiwei, Yang Jianhua, Chen Peng(Yunnan Academy of Forestry)//Journal of Northeast Forestry University,2017,45(3):77-81.
The phylogenetic relationships among threeBhutanitisspecies distribution in China areas were analyzed based on 658 bp sequences of COI and 480 bp ND1 gene. With COI gene sequences, the average T contents ofB.thaidina,B.mansfieldiandB.lidderdaliiof were 41.5%, 40.0%, 41.9%, C were 15.2%, 15.8%, 14.3%, A were 29.3%, 30.7%,29.8%, and G were 14.0%,13.5%, 14.0%, respectively. There were 131 variable sites, in which 56 were parsimony information sites, accounting for 19.91% and 8.51% of the total length, respectively. With the ND1gene sequences, T were 34.4%, 33.3%, 32.1%, C were 11.3%, 11.9%, 12.1%, A were 45.6%, 46.7%, 47.1%, and G were 8.7%, 8.1%, 8.8%. There were 63 variable sites, in which 39 were parsimony information sites, accounting for 13.13% and 8.13% of the total length, respectively.Bhutanitisexhibited a high A+T bias. A low level of genetic diversity in the total population ofBhutanitis, and their average genetic distance was small. From the phylogenetic trees NJ and BI,B.mansfieldiwas differentiated earlier than other species followed byB.lidderdalii.B.lidderdaliishowed the highest affinity withB.thaidinaandB.mansfieldi.
