应用R语言MetaOmics分析流程实现基因芯片数据的Meta分析
2017-07-20刘小平李晓东尹晓红刘新灿
刘小平,李晓东,郭 娇,曹 越,尹晓红,刘新灿
1. 武汉大学中南医院 循证与转化医学中心/武汉大学第二临床学院 循证医学与临床流行病学教研室/武汉大学循证与转化医学中心,武汉 430071; 2. 河南大学淮河医院 循证医学中心/科教科/泌尿外科,河南 开封 475000; 3. 河南中医药大学第一附属医院 心内科/河南中医药大学 循证医学中心,郑州 450000
应用R语言MetaOmics分析流程实现基因芯片数据的Meta分析
刘小平1,李晓东2,郭 娇3,曹 越1,尹晓红1,刘新灿3
1. 武汉大学中南医院 循证与转化医学中心/武汉大学第二临床学院 循证医学与临床流行病学教研室/武汉大学循证与转化医学中心,武汉 430071; 2. 河南大学淮河医院 循证医学中心/科教科/泌尿外科,河南 开封 475000; 3. 河南中医药大学第一附属医院 心内科/河南中医药大学 循证医学中心,郑州 450000
随着基因芯片技术的发展和检测成本的下降,近年来成千上万的基因表达谱研究被公开发表,因此基于基因表达芯片数据的Meta分析对于进一步验证具有重要意义。美国匹兹堡大学研究人员开发的基于R语言的MetaOmics分析流程目前包含的MetaQC、MetaDE和MetaPath程序包,可分别用于基因芯片数据Meta分析质量控制、差异基因筛选以及后续功能分析。本文以9例前列腺癌基因表达谱研究为例,介绍通过MetaOmics基因芯片数据实现Meta分析的具体步骤,为其临床应用提供参考。
Meta分析;基因芯片数据;MetaOmcis分析流程;R语言
微阵列(microarray)技术,又称基因芯片技术[1-2],主要是指由成千上万个DNA样品或寡核苷酸密集排列于硅片、玻片、聚丙烯或尼龙膜等固相支持物上,再与模板在严格条件下进行杂交,最后由激光共聚焦显微镜等设备获取图像信息,通过计算机分析处理获得信息的技术集合。基因芯片又常称为微排列或微点阵,目前新兴的DNA芯片(DNA chip)只是基因芯片中最主要的一种。根据固相载体上排列的探针不同,基因芯片可大致分为两类:①cDNA芯片,是将cDNA固定于玻片等固相载体表面,并将其暴露于一种或一组标记的探针之下,也可用基因组DNA或经过基因组错配扫描纯化的DNA来制造基因芯片;②寡核苷酸芯片,可通过原位合成法制备,亦可合成后再固定于芯片上,然后与标记的探针进行杂交[3]。近年来,由于基因芯片技术的发展和基因芯片检测成本的不断下降,国内外学者已经完成人体各种组织的基因表达谱检测[4]。然而,不同表达谱研究间的结论不尽相同。因此,基于基因表达谱数据的Meta分析,将荟萃分析不同基因芯片研究结果,以期扩大研究样本量、得出更为稳定的研究结论。
基于R语言的 MetaOmics是由美国匹兹堡大学的Tseng等开发的一组可用于基因芯片数据的Meta分析流程[5]。MetaOmics目前共包括MetaQC[6]、MetaDE[7-10]和MetaPath[11]三个R语言程序包,MetaQC用于基因芯片数据Meta分析数据前期质控,MetaDE程序包用于寻找差异基因,MetaPath主要用于后期功能富集分析。本文以前列腺癌数据基因表达数据集为例,介绍通过MetaOmics基因芯片数据实现Meta分析的具体步骤。
1 数据准备
1.1 基因表达谱数据的预处理和差异基因的筛选、注释
基因芯片数据一般是探针水平数据(杂交信号),基因芯片数据的预处理即是将探针水平数据转换成基因表达数据。表达谱数据质量控制的主要步骤包括背景校正(Background correction)、标准化(Normalization)、汇总(Summarization)及过滤(Filtering)[12]。背景校正主要用于消除非特异杂交和激光扫描过程中产生的噪音。目前比较流行的背景校正算法包括RMA(robust-multi-array average)和MAS 5.0等。标准化主要用于检测及校正探针信号值总分布的系统差异以及为不同来源的芯片数据的比较奠定基础。目前比较常见的标准化方法包括Cyclic loess、Quantile、Scaling、Nonlinear和VSN(variance stabilization and normalization)等。汇总指的是使用一定的统计学方法将之前得到的荧光强度值从探针水平汇总到探针组(probeset)水平。目前常用的归一化方法主要包括Li-Wong法、Median polish法和Affymetrix公司的汇总方法。过滤指的是去除一些基因,比如在各样本中表达值的变异相对较低的一些基因。R/bioconductor提供有Affy[13]、Simpleaffy[14]及AffyPLM[12]等多种程序包以实现基因芯片数据的Meta分析。
经过预处理后,需要对预处理后的数据进行一定的统计学分析,以得出特定条件下的显著性差异表达的基因。目前,相关的统计学方法主要包括变化倍数(fold-change,FC)标准、常规T检验、SAM(significance)法、方差分析(The Analysis of Variance, ANOVA)、经验贝叶斯法和RP(Rank Products)法等。R/bioconductor limma程序包提供了多种功能进行差异基因分析[15]。
得出差异基因后,常常需要对差异基因进行注释(annotation)。所谓的注释指的是,将探针组的每个探针映射到相应基因的国际标准名称(gene symbol)。目前,能够实现注释的工具包括Affymetrix公司提供的注释文件(http://www.affymetrix.com/support/technical/annotationfilesmain.affx)、基于R/bioconductor的annotate程序包(https://www.bioconductor.org/packages/3.3/data/annotation/),以及一些在线工具,如IDconverter(https://www.ncbi.nlm.nih.gov/pmc/utils/idconv/v1.0/)、DVAID(https://david.ncifcrf.gov/)等。
1.2 进一步准备基因表达的数据
基因芯片Meta分析的一大难点就是,前期数据的准备和将准备好的数据读入R软件中。用于Meta分析的基因芯片数据集主要涵盖以下内容:基因表达数据;结局变量,如疾病状态;病人特异性协变量,包括既往史以及相关临床和人口学信息等。将基因表达数据读入R之前,首先需要将其在Excel中整理成Metaomics能够识别的形式,支持两种类型的基因表达数据矩阵:①未经匹配的基因表达矩阵(图1),探针编号未映射到相应的基因名称(gene symbol),第1列是探针编号,第2列是相应的基因名称,第1行指的是样本编号,第2行指的是结局变量,剩余的是基因表达值的矩阵;②经匹配后的基因表达矩阵(图2),每一个基因都是唯一的编号,第1列即基因名称,第1行是样本编号,第2行指的是患者的生存期,第3行是删失值,其余的都是基因表达值的矩阵。
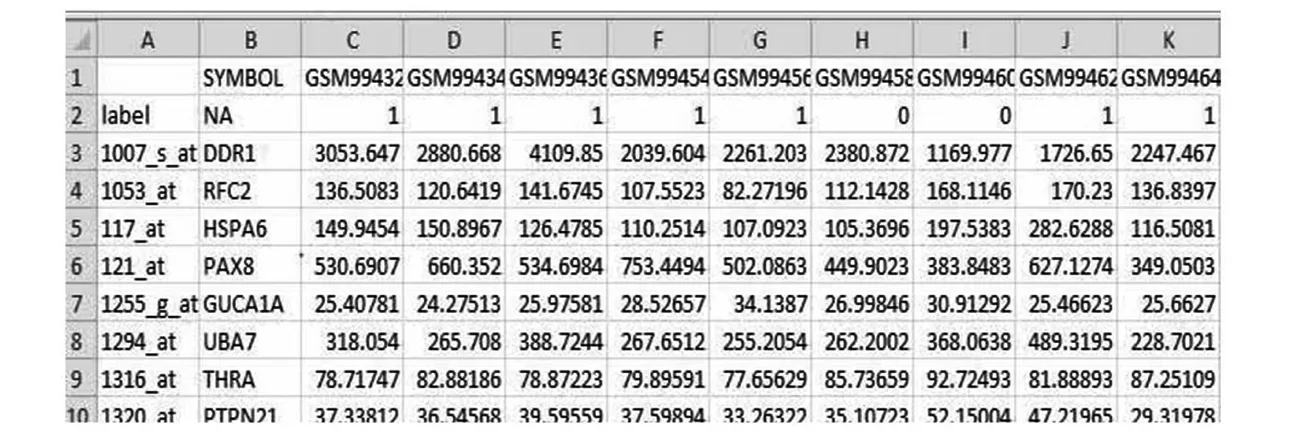
图1 未经匹配的基因表达矩阵
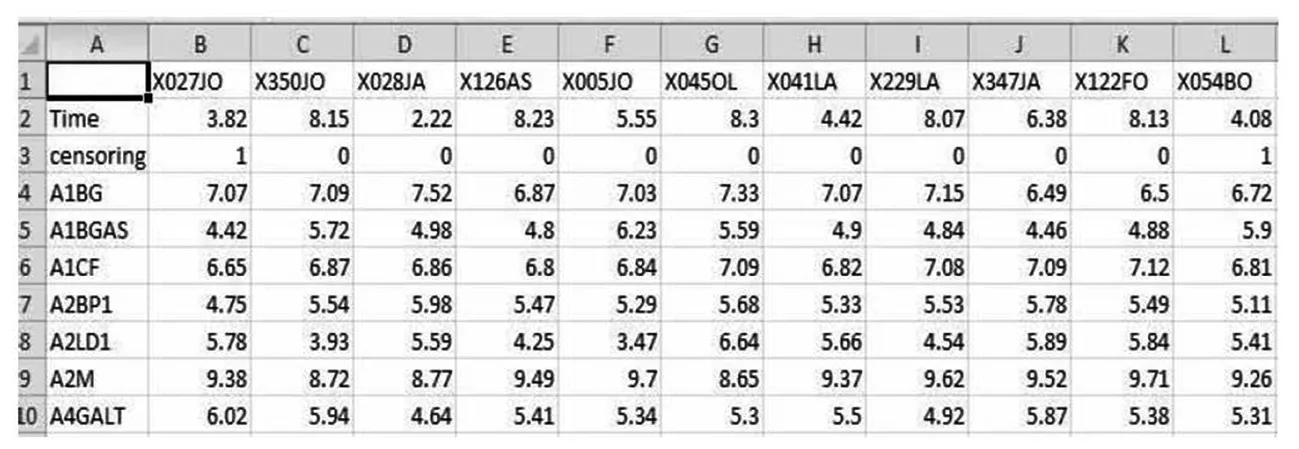
图2 经匹配后的基因表达矩阵

表1 9例前列腺癌数据集的一般情况
1.3 示例数据集
这里以表1所示的9例前列腺癌数据集为例,介绍使用MetaOmics实现基因芯片数据的Meta分析的流程。
2 软件的安装及相关程序包的加载
从R语言官方网站(https://www.r-project.org/)下载最新版R软件R-3.3.2。安装成功后,通过在R软件终端键入以下代码安装本次分析所需的程序包MetaDE和MetaQC:
install.packages(c(“MetaDE”,“MetaQC”))
同时,还需要在bioconductor安装运行MetaOmics所需要的impute程序包,具体代码如下:
source(“https://bioconductor.org/biocLite.R”)
biocLite(“impute”)
接着,可在R软件终端键入以下代码加载以上两个程序包:
library(MetaDE)
library(MetaQC)
至此,本次分析所需的程序包加载完毕。
3 完成Meta分析
3.1 数据读入
可通过以下代码,将本次分析的9例已经整理好的前列腺癌基因表达数据读入R软件:
study.names prostate.raw<-MetaDE.Read(study.names,skip=rep(1,9),via=“txt”,matched=T,log=F) 3.2 基因合并 由于多个表达数据集中的基因数目可能存在不一致的情况,MetaDE程序包提供了MetaDE.Merge函数,以在不同研究中提取出共同的基因供后续分析。此过程称为基因合并,具体代码如下: prostate.merged<-MetaDE.merge(prostate.raw) dim(prostate.merged〔〔1〕〕〔〔1〕〕) 其输出结果为: 〔1〕 1903 34 # 通过基因合并,筛选出1 903个基因,它们共同存在于所纳入的9例前列腺癌基因表达谱中。 3.3 基因过滤 由于在生物学上许多基因是不表达或无意义的,在进行基因表达分析的过程中,这些基因的存在将会产生错误发现(false discovery),所以在进行Meta分析之前应先将样本间平均信号强度较低和标准差较低的基因过滤掉。这里,我们剔除平均信号强度和标准差前30%的基因,具体代码如下: prostate.filtered<-MetaDE.filter(prostate.merged,c(0.3,0.3)) dim(prostate.filtered〔〔1〕〕〔〔1〕〕) 3.4 质量控制 MetaQC程序包提供6种定量质量控制指标,包括研究间共表达框架的内部同质性(即内部质控,internal quality control,IQC)、通路数据库相关共表达框架的外部一致性(即外部质控,external quality control,EQC)、差异基因检测的准确性(AQCg)、通路检测的准确性(AQCp)、差异表达基因排序一致性(CQCg)、通路排序的一致性(CQCp)。同时,MetaQC程序包还可以进行主成分分析(principal component analysis,PCA),以进一步将质控结果进行可视化。MetaQC函数以定量实现质量控制,具体的质控步骤可分为以下五步。 第一步、生成QC对象。具体代码如下: Data.QC<-list() for(i in 1:9){ colnames(prostate.filtered〔〔i〕〕〔〔1〕〕)<-prostate.filtered〔〔i〕〕〔〔2〕〕 Data.QC〔〔i〕〕<-impute.knn(prostate.filtered〔〔i〕〕〔〔1〕〕)$data print(dim(Data.QC〔〔1〕〕)) } names(Data.QC)<-names(prostate.filtered) ProstateQC<-MetaQC(Data.QC,“c2.all.v5.2.symbols.gmt”,filterGenes=F,verbose=TRUE,isParallel=TRUE,nCores=12) 第二步、使用runQC函数对OC对象进行质量控制。其具体代码如下: runQC(ProstateQC,B=1e4,fileForCQCp=“c2.all.v5.2.symbols.gmt”) 第三步、使用print函数查看质控结果。其具体代码如下: print(ProstateQC) 输出结果,见图3。 图3 MetaQC质量控制输出结果 第四步、使用plot函数查看可视化结果。其具体代码如下: plot(ProstateQC) 输出结果见图4。 图4 9例前列腺癌基因表达矩阵质量控制指标主成分分析(PCA) 第五步、剔除不合格研究。根据metaQC结果,并考虑样本量、芯片平台等,我们剔除Nanni、Dhanasekaran和Tomlins三个研究,然后再次进行基因合并和基因过滤,以进行后续分析。其具体代码如下: study.names<-c(“Welsh”,“Yu”,“Lapointe”,“Varambally”,“Singh”,“Wallace”) data.QC.raw<-list() for(i in 1:length(study.names)){ data.QC.raw〔〔i〕〕<-prostate.raw〔〔study.names〔〔i〕〕〕〕 } names(data.QC.raw)<-study.names data.QC.merged<-MetaDE. merge(data.QC.raw) data.QC.filtered<-MetaDE.filter(data.QC.merged,c(0.3,0.3)) 3.5 差异表达分析 MetaDE程序包提供三类函数实现基因芯片数据的Meta分析,包括MetaDE.rawdata(基于原始数据的meta分析)、MetaDE.pvalue(基于P值的Meta分析)、MetaDE.ES(基于效应量Meta分析)。其中,各研究的P值和效应量(effect size)可分别用ind.analysis和ind.cal.ES计算,也可有第三方软件计算。本例由于原始数据可得,我们使用Meta.rawdata实现Meta分析,具体代码如下: MetaDE.Res<-MetaDE.rawdata(data.QC.filtered,ind.method=rep(“modt”,6),meta.method= c(“Fisher”,“maxP”),rth=4,nperm=300,asymptotic=F) 同时,MetaDE程序包可通过heatmap.sig.genes函数绘制基于特定的P值或FDR值差异表达基因热图。heatmap.sig.genes也可以表格的形式显示差异表达基因。具体代码如下: label1<-rep(0:1,each=5) label2<-rep(0:1,each=5) exp1<-cbind(matrix(rnorm(5*200),200,5),matrix(rnorm(5*200,2),200,5)) x<-list(list(exp1,label1),list(exp2,label2)) heatmap.sig.genes(MetaDE.Res,meta.method=“maxP”,fdr.cut=0.00001,color=“GR”)# 图5 经Meta分析后的差异表达基因热图 输出结果见图5。为了便于可视化,本次分析将fdr.cut设置为0.000 01,即错误发现率小于0.000 01。实际分析中可根据具体需要调整。 近年来,随着基因表达谱研究的普及,越来越多的基因表达数据被共享。Meta分析作为一种二次分析方法,能够权衡各研究中的结果,扩大研究样本量,最终得出稳定、可靠的研究结论[16-17]。因此,基于基因表达谱数据的Meta分析研究也具有重要意义[18]。 如前所述,MetaOmics提供MetaQC进行基因表达矩阵的质量控制,MetaDE进行差异基因的筛选,MetaPath进行后续功能富集分析。因当前MetaPath分析过程相对复杂,而且有较多工具可进行基于差异基因的功能富集分析,如DAVID(https://david.ncifcrf.gov/),KEGG数据库(http://www.kegg.jp/)等,所以本文不就MetaPath进行叙述。 在进行基因芯片数据Meta分析时,也有一些注意事项。由于采用整段目的基因序列设计探针会导致非特异性结合和噪声,所以设计探针时,一般采用目的基因中一些短的、高特异性基因。不同的设计标准导致多个探针映射到相同的基因。由于探针序列的特异性不够,可能出现某一探针被映射到多个基因ID的情况,也可能出现某些探针并不能映射到任何基因ID的情况,因此研究人员需要在某一基因芯片数据集内部和不同的数据集间确定是哪一组探针代表某一指定基因。有研究认为,当多个探针映射到相同的基因时,选择四分位间距最大的探针比较合适[1]。此外,如同其他类型的Meta分析,为保证研究结果的可靠性,基于基因芯片数据的Meta分析研究同样应当严格遵循相关报告规范[19]。 [1] Jaksik R, Iwanaszko M, Rzeszowska-Wolny J, et al. Microarray experiments and factors which affect their reliability [J]. Biol Direct, 2015,10:46. [2] Lévêque N, Renois F, Andréoletti L. The microarray technology: facts and controversies [J]. Clin Microbiol Infect, 2013,19(1):10-14. [3] Miller M B, Tang Y W. Basic concepts of microarrays and potential applications in clinical microbiology [J]. Clin Microbiol Rev, 2009,22(4):611-633. [4] Rung J, Brazma A. Reuse of public genome-wide gene expression data [J]. Nat Rev Genet, 2013,14(2):89-99. [5] Wang X, Kang D D, Shen K, et al. An R package suite for microarray meta-analysis in quality control, differentially expressed gene analysis and pathway enrichment detection [J]. Bioinformatics, 2012,28(19):2534-2536. [6] Kang D D, Sibille E, Kaminski N, et al. MetaQC: objective quality control and inclusion/exclusion criteria for genomic meta-analysis [J]. Nucleic Acids Res, 2012,40(2):e15. [7] Li J, Tseng G C. An adaptively weighted statistic for detecting differential gene expression when combining multiple transcriptomic studies [J]. Annals of Applied Statistics, 2012,5:994-1019. [8] Lu S, Li J, Song C, et al. Biomarker detection in the integration of multiple multi-class genomic studies [J]. Bioinformatics, 2010,26(3):333-340. [9] Wang X, Lin Y, Song C, et al. Detecting disease-associated genes with confounding variable adjustment and the impact on genomic meta-analysis: with application to major depressive disorder [J]. BMC Bioinformatics, 2012,13:52. [10] Tseng G C, Ghosh D, Feingold E. Comprehensive literature review and statistical considerations for microarray meta-analysis [J]. Nucleic Acids Res, 2012,40(9):3785-3799. [11] Shen K, Tseng G C. Meta-analysis for pathway enrichment analysis when combining multiple genomic studies [J]. Bioinformatics, 2010,26(10):1316-1323. [12] Bolstad B M. Low-level analysis of high-density oligonucleotide array data: background, normalization and summarization [D]. Berkeley: University of California, 2004. [13] Gautier L, Cope L, Bolstad B M, et al. Affy-analysis of Affymetrix GeneChip data at the probe level [J]. Bioinformatics, 2004, 20(3): 307-315. [14] Wilson C L, Miller C J. Simpleaffy: BioConductor package for Affymetrix quality control and data analysis [J]. Bioinformatics, 2005,21(18):3683-3685. [15] Ritchie M E, Phipson B, Wu D, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies [J]. Nucleic Acids Res, 2015,43(7):e47. [16] 曾宪涛,田国祥,张超,等. Meta分析系列之十五:Meta分析的进展与思考 [J]. 中国循证心血管医学杂志,2013,5(6):561-563,587. [17] 曾宪涛. 再谈循证医学 [J]. 武警医学,2016,27(7):649-654. [18] Walsh C J, Hu P, Batt J, et al. Microarray Meta-Analysis and Cross-Platform Normalization: Integrative Genomics for Robust Biomarker Discovery [J]. Microarrays (Basel), 2015,4(3):389-406. [19] Ramasamy A, Mondry A, Holmes C C, et al. Key issues in conducting a meta-analysis of gene expression microarray datasets [J]. Plos Med, 2008,5(9):184. [责任编辑 段金卯] Application Meta Analysis of Microarray Studies Using the R pipeline MetaOmics LIU Xiaoping1, LI Xiaodong2, GUO Jiao3, CAO Yue1, YIN Xiaohong1, LIU Xincan3■ 1. Center for Evidence Based and Translational Medicine, Zhongnan Hospital of Wuhan University/Center for Evidence Based and Translational Medicine, Wuhan University/Department of Evidence Based Medicine and Clinical Epidemiology, The Second Clinical Medical College of Wuhan University, Wuhan 430071, China; 2. Center for Evidence Based Medicine/Management Office of Scientific Research and Postgraduate Affairs/Department of Urology, Huaihe Hospital of Henan University, Kaifeng 475000, China; 3. Department of Cardiology, First Affiliated Hospital/Center for Evidence Based Medicine, Henan University of Traditional Chinese Medicine, Zhengzhou 450000, China With the development of microarray and the decrease of its associated detection cost, thousands of microarray studies have been publicly published in recently years, and meta analysis based on microarray data might be of great significance for further verification. The R pipeline MetaOmics, published by researchers from Pittsburgh University, US, contains three different R packages (MetaQC, MetaDE, and MetaPath), which could be applied to conducting quality control, the identification of differentially expressed genes, and its corresponding downstream enrichment analysis, respectively. In the present study, we introduced the workflows of MetaOmics to conduct a meta analysis of microarray data using gene expression data of 9 different microarray studies. Meta-analysis; microarray data; MetaOmics pipeline; R language 1672-7606(2017)02-0123-06 2017-02-21 河南省中西医结合心血管疑难危急重症创新团队项目 刘小平(1989-),男,湖北孝感人,研究实习员,研究方向:循证医学。 ■通信作者:刘新灿(1965-),男,河南新密人,主任医师、教授,研究方向:循证心血管病学及心血管疾病的诊治。E-mail:liuxincan103@163.com。 R-39 C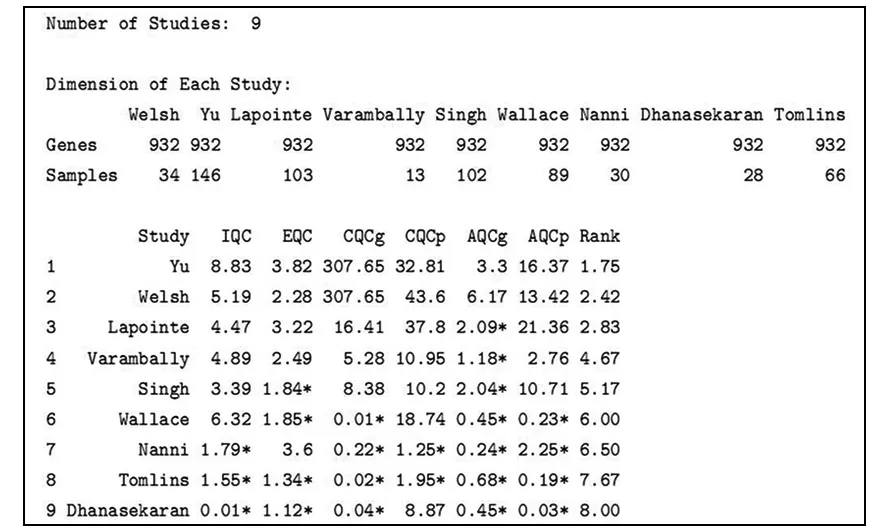


4 讨论
