Ni掺杂TiO2电子和几何结构的第一性原理研究
2017-07-12于智清薛向欣
于智清,王 逊,杨 合, 薛向欣
(1. 东北大学冶金学院,沈阳110819;2. 沈阳建筑大学理学院,沈阳110168)
Ni掺杂TiO2电子和几何结构的第一性原理研究
于智清1, 2,王 逊2,杨 合1, 薛向欣1
(1. 东北大学冶金学院,沈阳110819;2. 沈阳建筑大学理学院,沈阳110168)
利用密度泛函理论框架下的第一性原理方法,对不同掺杂量、不同方式的Ni掺杂锐钛矿相TiO2(anatase TiO2:A-TiO2)的晶胞结构、缺陷形成能和态密度图像等进行了研究,并着重讨论了掺杂Ni离子周围配位场改变对体系电子结构的影响.结果表明,掺杂方式的不同对Ni-TiO2超晶胞的几何结构有较大影响.正是几何结构的变化导致掺杂Ni离子处于不同类型的晶体配位场之中,这是掺杂体系电子结构差异的本质原因.通过对掺杂体系形成能的比较,发现Ni掺杂TiO2的具体方式取决于晶体生长过程中的氧环境.通过对体系电子结构的研究,发现各种掺杂体系的禁带中都出现由Ni 3d-O 2p轨道杂化形成的杂质能级.Ni离子替代晶格Ti掺杂会使吸收光谱红移,而Ni的晶隙掺杂则使吸收光谱蓝移,且这种光谱吸收带边的差异会随掺杂浓度的增加而增大.
Ni掺杂;掺杂方式;晶胞结构;态密度;第一性原理
TiO2是一种优良的半导体材料,具有储存量大、价格合理和稳定性高等优势,广泛地应用在有机污染物处理、清洁能源制造、表面抗菌、金属防腐等诸多领域[1].在以上各种应用过程中,都需要TiO2材料能够有效地吸收光子以产生大量的光生(e-/h+)对,并使产生的光生载流子在其生命周期内顺利地迁移到晶体表面[2].但是,本征TiO2的禁带宽度较大(3.0~3.2 eV),使其只能吸收部分紫外光线的光子(波长λ<380 nm),且光生(e-/h+)对的复合率较高,这些都严重地限制了TiO2材料的实际应用[3].
实验和理论研究都表明,利用过渡金属元素对本征TiO2进行掺杂,可以使体系的光吸收带边红移,从而提升光子吸收效率.比如,实验发现Fe、Mo、Ru、Os、Re、V和Rh等多种离子掺杂都可以提高体系的催化效率[4-8].理论研究认为,这是因为金属离子的掺杂可以在TiO2禁带之中引入杂质能级,并且杂质能级在禁带之中的深度与掺杂离子的价电子数有关[9].另外,掺杂的金属离子使晶胞结构产生畸变并进一步影响体系的能带结构,最终使TiO2光吸收带边发生红移.
近年来,实验方面对不同的金属离子掺杂TiO2体系性能进行了大量的比对研究.这些工作发现Ni掺杂不仅可以提高掺杂体系的可见光利用率和催化效率,还具有掺杂体系易形成、稳定性较高、无毒性和导带电子还原能力强等优点[10].
理论方面也对Ni-TiO2掺杂体系开展了一些研究[5,9,11-12],但还不够全面.比如,M. Sun等[10]通过实验证明,Ni进入TiO2晶格有替Ti位掺杂和晶隙掺杂两种方式.但理论研究中一直忽视了Ni离子晶隙掺杂对于体系的影响.此外,理论研究也未对Ni-TiO2晶胞的体积变化等问题给予合理的解释[7,13].这些都需要更加全面的研究和讨论.为此,本文将使用密度泛函理论框架下的第一性原理方法,系统地分析不同掺杂方式的Ni掺杂所导致的晶体场类型变化对TiO2体系性能的影响,以期对具体的实验过程有所帮助.
1 计算方法
本文计算采用自旋密度泛函理论(Spin density functional theory:SDFT)框架下的平面波超软赝势法.在能量计算过程中,由于DFT方法在求解Kohn-Sham方程时,没有充分考虑d/f轨道电子的交换关联能,所以传统的局域密度近似(LDA)和广义梯度近似(GGA)方法在计算以过渡金属氧化物为代表的强关联体系时,计算结果与实验偏差都很大.特别是在计算杂质能级位置时,往往同实验规律相背离[14-15].研究表明,利用广义梯度近似加排斥能修正(GGA+U)的方法可以大幅地提高计算精度[14-16].比如,在计算含氧空位锐钛矿相TiO2的能带结构时,传统的GGA方法所得的杂质能级紧邻导带底部,而实验发现此杂质能级距离导带底0.8~1 eV.Cheng[15]和Mattioli[16]等分别利用GGA+U的方法得到了与实验值完全吻合的结果.所以,本文也将采用GGA+U(PBE)的方法对体系进行计算.为取得更好的计算效果,Hunnard U的取值为文献推荐值UTi=4 eV,UNi=3.5 eV[17-18].
TiO2具有锐钛矿、金红石和板钛矿三种晶型.其中使用最广泛的是锐钛矿(A-TiO2)[19].每个A-TiO2晶胞包含4个Ti原子和8个O原子.用Ni离子替换晶格Ti离子或置于A-TiO2晶格空隙之中来模拟Ni替位掺杂NiTi和晶隙掺杂Niin,如图1(a)、(b)所示.为模拟的不同Ni掺杂量,分别构建了2×2×1和2×2×2 A-TiO2超晶胞.Ni掺杂量(摩尔分数)为Niin(1.03%、2.04%),NiTi(1.04%、2.08%),此掺杂量在实验中可实现,也不会产生Ni团簇等现象[9-10,13].倒空间k网格设置为4×4×4 (2×2×1超晶胞) 和4×4×2 (2×2×2超晶胞)[20]. 设置平面波截断能量为400 eV.设置自洽场收敛标准为5×10-7eV/atom.所有计算使用CASTEP软件完成.
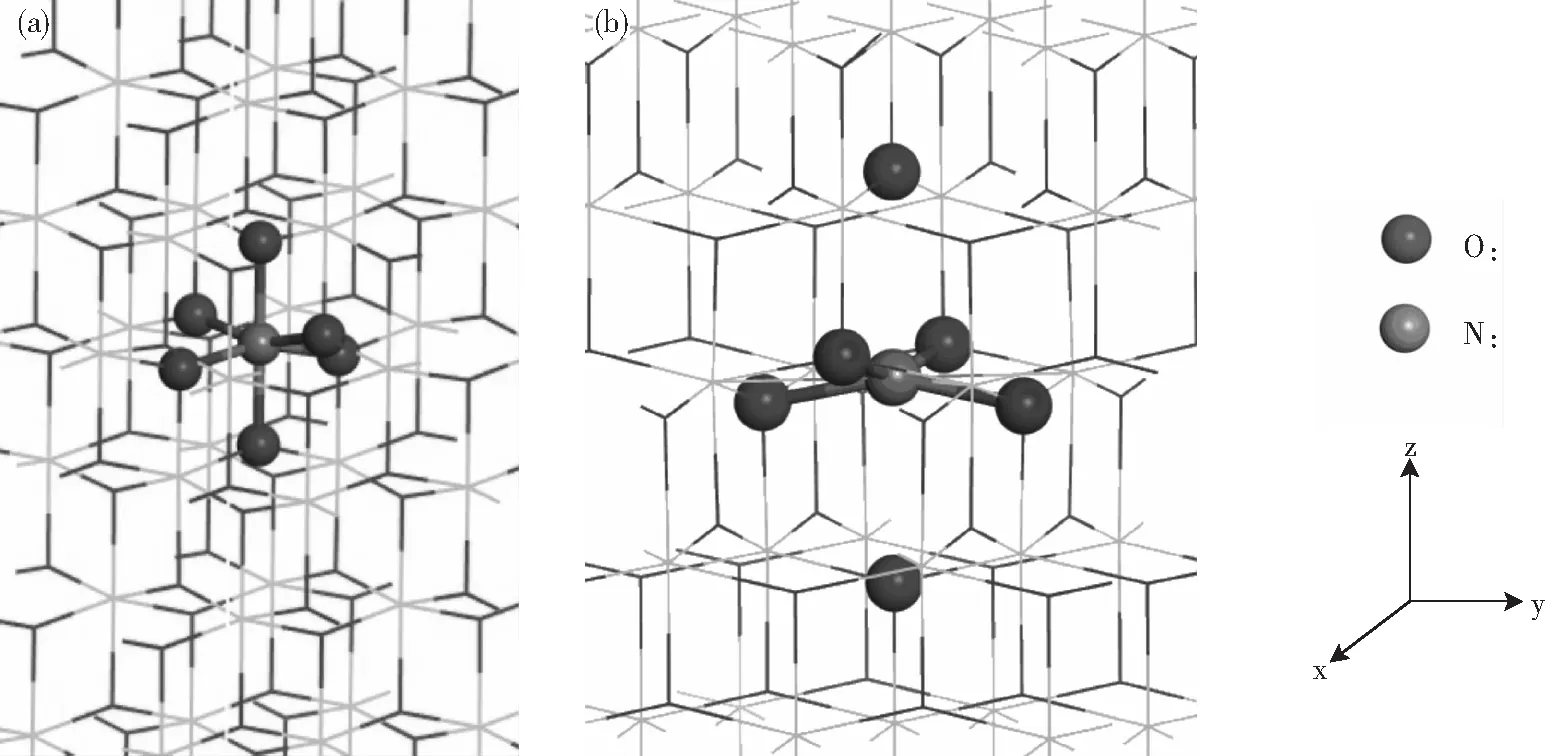
图1 Ni掺杂TiO2超晶胞(NiTi;Niin)Fig.1 Structures of the nickel doped A-TiO2: (a)—NiTi ; (b)—Niin
2 计算结果分析
2.1 几何结构分析
在本征A-TiO2晶胞中,Ti离子与其近邻的O离子组成体心八面体结构的TiO6.八面体中Ti-O键的长度分别为deq和dap.几何结构计算结果如表1.比对计算得到的A-TiO2单胞几何参数与实验报道值,发现最大偏差在3%以内,可以保证计算合理、有效.
根据表1中数据,Ni替代Ti位掺杂时(NiTi):新形成的NiO6八面体中的6条Ni-O键的长度均小于对应的Ti-O键长,NiO6八面体的体积也小于TiO6八面体.说明八面体6个顶点上的O离子距离Ni离子更近,这会使O离子形成的配位场增强,加大中心离子的轨道分裂.同时,所成Ni-O键的布居值(0.29,0.23)也远小于对应的Ti-O键(0.71,0.29),这说明掺杂体系的共价性降低和系统稳定性下降.根据实验测定,Ni2+的离子半径(0.069 nm)应大于Ti4+的离子半径(0.060 5 nm)[10].但是,掺杂形成的Ni-O键长却小于对应的Ti-O键长和NiO晶体的Ni-O键长(0.209 4 nm).Ni-O键长度的减小和离子性的增强都意味着NiTi掺杂时,Ni离子的价态较高Nix+(x>2).此外,晶胞体积的变化也说明,替位掺杂量越高,超晶胞的体积随掺杂量的升高而降低越小.此时晶粒体积减小、比表面积增加、量子效应增强、粒子的表面活性也会提高[3].
当Ni离子采取晶隙掺杂时(Niin):晶格间隙中的Ni离子与最近邻的4个O原子成键,而与2个z方向的第二近邻O离子距离太远不能成键,形成稍微扭曲的NiO4平面正方形.此时,中心的Ni离子受平面正方形配位场作用.所成的4个 Ni-O 键的键长小于NiTi掺杂所成Ni-O键,但仍小于NiO晶体中的Ni-O键.说明此时的掺杂Ni离子的价态应低于NiTi掺杂时Ni离子的价态.由于Niin掺杂的过程中,额外加入的Ni离子使体系电子浓度增加,晶体内部库伦排斥力增强,晶胞体积变大,晶体稳定性随之降低.综合两种掺杂情况, Ni掺杂的不同方式对Ni离子周围的化学环境有很大影响,但掺杂量的改变对体系影响不大,说明此掺杂量范围的掺杂Ni离子之间没有产生较强的相互作用.
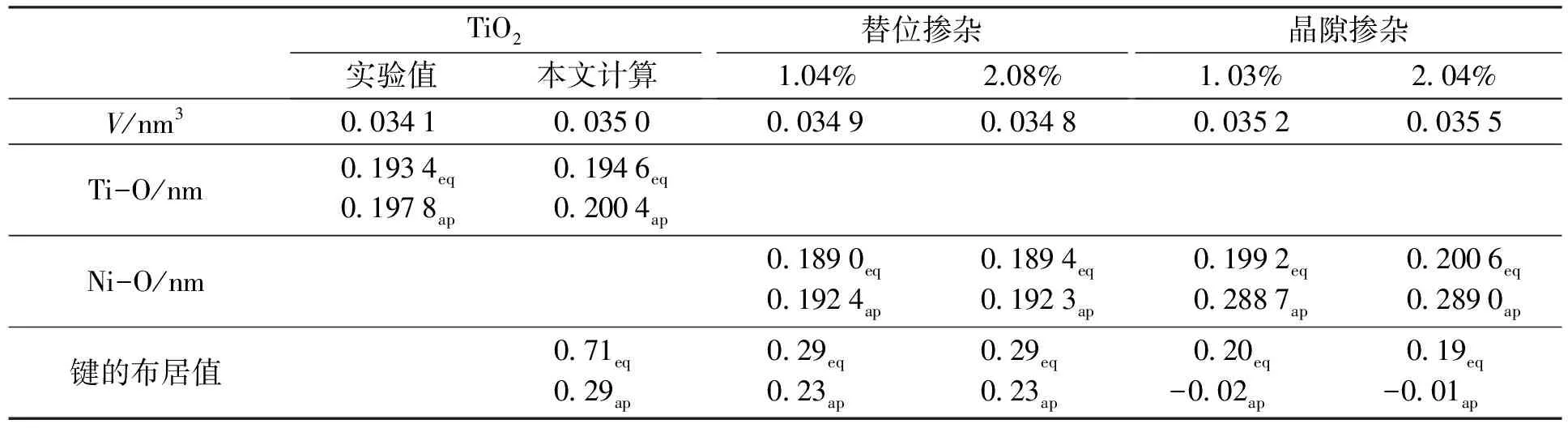
表1 几何结构计算结果
2.2 形成能分析
形成能计算是比较不同体系热稳定性差异的重要手段.本文使用公式(1)[20]对掺杂体系的形成能进行了计算,所得结果如表2.
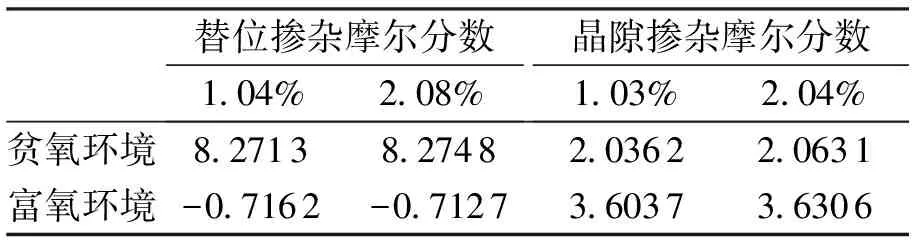
表2 掺杂体系形成能(eV)
Eform=Edoped-Epure-μNi+μTi
(1)
Eform、Epure、Edoped分别为Ni掺杂体系的形成能以及纯TiO2超晶胞和掺杂体系的系统总能量.μNi、μTi为Ni原子和Ti原子的化学势.化学势取值与掺杂体系的生长氧环境有关.晶体在贫氧环境中生长时(O-poor),μNi、μTi通过计算Ni、Ti金属块体的能量得到.μO晶体在富氧环境下生长时(O-rich),μNi、μTi通过公式(2)、(3)计算得到.μO通过计算氧气中的一个氧原子的能量得到,μO=1/2μ(O2).
2μO+μTi=μ(TiO2)
(2)
μO+μNi=μ(NiO)
(3)
由表2可知:贫氧环境生长的晶体,掺杂Ni离子更倾向于进入TiO2超晶胞的晶隙之中,形成晶隙掺杂;而富氧环境生长的晶体,掺杂Ni离子更易于取代晶格Ti离子,形成替Ti位掺杂.相对于晶隙掺杂,替位掺杂的形成能更容易受氧环境的影响.尤其是在富氧环境下,替位掺杂的形成能已经变为负值,说明替位掺杂易形成且产物稳定性较高.形成能计算表明,可以通过改变掺杂体系生长过程中的氧环境来选取不同的Ni掺杂方式,这将有利于具体实验的操作.
2.3 态密度分析
为研究Ni掺杂对本征TiO2体系电子结构的影响,绘制了不同Ni-TiO2掺杂体系的态密度图像.图2(a)表明:本征TiO2作为典型的金属氧化物半导体材料,其费米能级(虚线)位于价带顶部.禁带宽度计算值为2.445 eV.如图2(b)、(c),替位掺杂时,其费米能级位置没有变化,仍旧位于价带顶,这时的禁带宽度Eg大于纯TiO2,禁带宽度随掺杂浓度增加而略有加宽.Ni 3d电子轨道和O 2p轨道杂化,在禁带中形成两条杂质能级(价带顶之上0.94~1.37 eV处).这说明掺杂的 Ni 3d轨道能量低于被替换的Ti 3d轨道;同时也说明,掺杂Ni离子周围的配位O原子的能级发生了分裂.因为杂质能级的位置距离价带顶部和导带底部都很远,且其电位高于费米能级,所以导带电子和激发的价带电子都会将优先回落或激发到杂质能级.这就是掺杂体系吸收带边红移的根本原因[5].
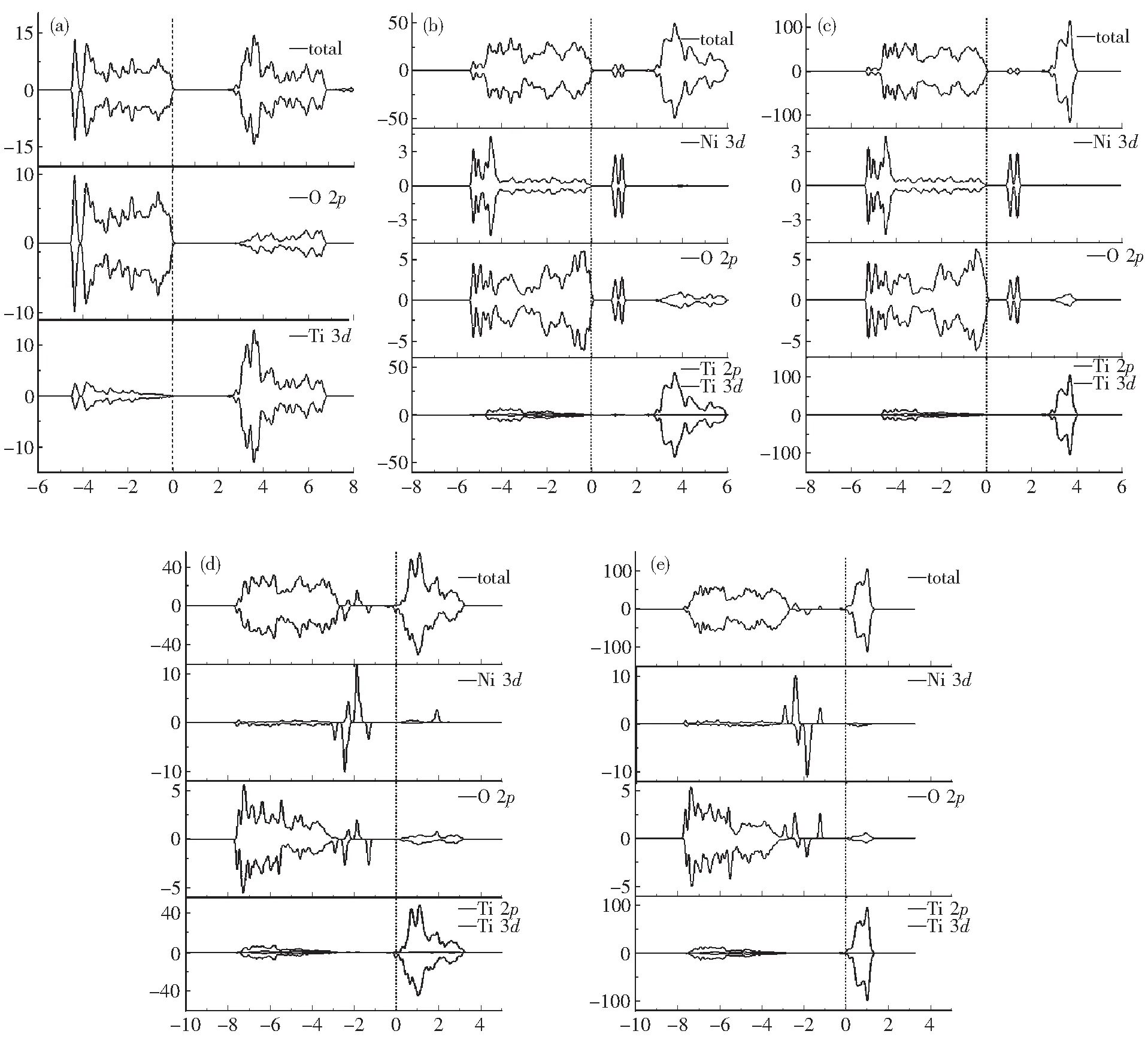
图2 纯TiO2和Ni掺杂体系电子态密度图Fig.2 Density of electronic states for the pure TiO2 and nickle doped system:(a)—纯TiO2; (b)—NiTi1.04%; (c)—NiTi2.08%; (d)—Niin1.03%; (e)—Niin2.04%
如图2(d)、(e),当Ni离子采取晶隙掺杂时:相对本征TiO2,掺杂体系的禁带宽度有所减小.此时,掺杂Ni离子将多余电子引入体系,促使费米能级电位升高并进入导带,体系呈现明显的金属性.多条杂质能级出现在禁带之中(高于价带顶部0.26~1.43 eV处).需要说明的是,这些禁带中的杂质能级都在费米能级以下.依据能带填充原理,费米能级以下的电子态为占据态,价带电子只能从价带顶向费米能级以上的非占据态能级跃迁[3],完成跃迁需要吸收更大能量的光子才能进行. 所以,晶隙掺杂时,光吸收带边会因此发生蓝移现象.同时,替位掺杂使部分本为空的Ti 3d能级被电子占据,导致Ti3+离子产生Ti4++e-=Ti3+.大量研究指出,Ti3+是高效的e-/h+分离中心和离子活化中心,且电子在Ti 3d能级具有较小的有效质量[12-13],这些将对晶体催化效率和晶体光电性能的提升有重要意义.
3 结 论
(1)富氧的晶体生长环境有利于掺杂Ni离子进入TiO2超晶胞的晶格空隙之中形成晶隙掺杂;而贫氧的生长环境则有利于Ni离子替代Ti离子形成替位掺杂.相对于替位掺杂,晶隙掺杂的形成能更容易受晶体生长的氧环境影响,在富氧生长环境下,替位掺杂的形成能最低(-0.716 2 eV,-0.712 7 eV),说明此时很容易形成替位掺杂,并且掺杂产物的稳定性很高.
(2)替位掺杂时,形成的Ni-O键的长度小于对应的Ti-O键长.Ni-O键的平均布居值也小于被取代的Ti-O键,形成键的共价性较弱,最终导致晶体的稳定性下降.另外,随着掺杂量的升高,晶胞体积减小.晶隙掺杂的过程将向体系引入额外的电子,晶体场排斥作用增加,晶胞体积随之变大,晶体稳定性下降.
(3)Ni离子的替位掺杂将使禁带宽度增加,而晶隙掺杂会使禁带宽度减小.变化的幅度将随掺杂浓度的增加而增大.形成的杂质能级由Ni 3d和O 2p轨道的杂化而成.替位掺杂虽能使吸收光谱的带边红移,但也容易成为光生电子e-和空穴h+的复合中心.晶隙掺杂则会使吸收光谱带边蓝移.
[1]Gombac V, Rogatis L De, Gasparotto A,etal. TiO2nanopowders doped with boron and nitrogen for photocatalytic applications[J]. Chemical Physics, 2007, 339(1-3): 111-123.
[2]Geng H, Yin S W, Yang X,etal. Geometric and electronic structures of the boron-doped photocatalyst TiO2[J]. J Phys: Condens Matter, 2006, 18(1): 87-96.
[3]Yang K S, Dai Y, Huang B B. Origin of the photoactivity in boron-doped anatase and rutile TiO2calculated from first principles[J]. Phys Rev B, 2007, 76(19): 195201.
[4]Choi W Y, Termin A, Hoffmann M R. Effects of metal-ion dopants on the photocatalytic reactivity of quantum-sized TiO2particles[J]. J Phys Chem, 1994, 33(10):1091-1092.
[5]Zhao L, Park S G, Blanka M K,etal. Dopant selection rules for desired electronic structure and vacancy formation characteristics of TiO2resistive memory[J]. Appl Phys Lett, 2013, 102(8): 083506.
[6]Yao Z P, Jia F Z, Tian S J,etal. Microporous Ni-doped TiO2film photocatalyst by plasma electrolytic oxidation[J]. Applied Materials and Interfaces, 2010, 2(9): 2617-2622.
[7]Visinescu C M, Sanjines R, Levy F,etal. Photocatalytic degradation of acetone by Ni-doped titania thin films prepared by dc reactive sputtering[J]. Applied Catalysis B: Environmental 2005, 60:155-162.
[8]Pol R, Guerrero M, Garcia-Lecina E,etal. Ni-Pt- and (Ni/Pt)-doped TiO2nanophotocatalysts: A smart approach for sustainable degradation of Rhodamine B dye[J]. Applied Catalysis B: Environmental, 2016, 181:270-278.
[9]赵宗彦,柳菊清,张瑾,等. 过渡金属掺杂锐钛矿相TiO2的第一性原理研究[J]. 物理学报, 2007, 56(11):6592-6599. (Zhao X Y, Liu Q J, Zhang J,etal. First-principles study of 3d transition metal-doped anatase[J]. Acta Phys Sin, 2007, 56(11): 6592-6599.)
[10]Sun M M, Chen Z Y, Yu J Q. Highly efficient visible light induced photoelectrochemical anticorrosion for 304 SS by Ni-doped TiO2[J]. Electrochimica Acta, 2013, 109:13-19.
[11]张小超,赵丽军,樊彩梅,等. 过渡金属(Fe,Co,Ni,Zn)掺杂金红石TiO2的电子结构和光学性质[J]. 物理学报, 2012, 61(7):077101. (Zhang X C, Zhao L J, Fan C M,etal. Electronic structures and optical properties of transition metals(Fe,Co,Ni,Zn) doped rutile TiO2[J]. Acta Phys Sin, 2012, 61(7): 077101.)
[12]Chen J, Lu G H, Cao H H,etal. Ferromagnetic mechanism in Ni-doped anatase TiO2[J]. Appl Phys Lett, 2008 93(17):172504.
[13]Cho J H, Hwang T J, Joh Y G,etal. Room-temperature ferromagnetism in highly-resistive Ni-doped TiO2[J]. Appl Phys Lett 2006, 88(9):092505.
[14]Aryasetiawan F,Karlsson K,Jepsen O,etal. Calculations of hubbard U from first-principles[J]. Phys Rev B, 2006, 74(12):5106.
[15]Cheng H Z, Selloni A. Energetics and diffusion of intrinsic surface and subsurface defects on anatase TiO2(101) [J]. J Chem Phys, 2009 131(5): 054703.
[16]Mattioli G, Filippone F, Alippi P,etal. Ab initio study of the electronic states induced by oxygen vacancies in rutile and anatase TiO2[J]. Phys Pev B:Condens Matter Mater Phys , 2008, 78(24):1201.
[17]Ruiz Preciado M A, Kassiba A, Acevedo M,etal. Vibrational and electronic peculiarities of NiTiO3nanostructures inferred from first principle calculatons[J]. RSC Adv, 2015, 5(17):396-404 .
[18]Yang J, Lv C Q, Guo Y,etal. A DFT+U study of acetylene selective hydrogenation on oxygen defective anatase (101) and rutile (110) TiO2supported Pd4 cluster[J]. J Chem Phys, 2012, 136(10):104107.
[19]Luttrell T, Halpegamage S, Tao J,etal. Why is anatase a better photocatalyst than rutile——Model studies on epitaxial TiO2films [J]. Sci Rep, 2014, 4:4043.
[20]Yu J G, Zhou P, li Q,etal. New insight into the enhanced visible-light photocatalytic activities of B-, C- and B/C-doped anatase TiO2by first-principles[J]. Phys Chem Chem Phys, 2013, 15:12040-12047.
First principle study on the electronic and geometical structures of Ni-doped anatase TiO2
Yu Zhiqing1,2, Wang Xun2, Yang He1, Xue Xiangxin1
(1.School of Metallurgy, Northeastern University, Shenyang 110819, China; 2.School of Science, Shenyang Jianzhu University, Shenyang 110168, China)
With the first principle method of spin density functional theory, the crystal structure, formation energy of defect, and the density of states of anatase TiO2(A-TiO2), doped with different nickel content and different ways, were studied. Here, effect of change of the ligand field around the doping nickle ions on the electronic structure of the system was discussed. The results showed that, the difference of the doping ways has a great influence on geometric structures of the doped supercell, which makes change of the crystal ligand field around doping Ni ions. That is the essential reason for the difference of the electronic structure of the doped system. Compared the formation energy of the system, it was found that the doping ways depends on the oxygen environment in the crystal growth process. Through research on the electronic structure of the system it was found that the impurity band level is formed by the hybridization of Ni 3dand O 2porbitals. Replacement of the titanium crystal lattic by the nickel one causes red shift for the absorption spectrum. While the doped nickel results in blue shift for the spectrum. And furthermore, the difference of the absorption borderline will increase with the doped concentration.
doped anatase; doping mode; cell structure; density of states; first principle
10.14186/j.cnki.1671-6620.2017.02.013
O 775
A
1671-6620(2017)02-0154-05
