乙烯选择性齐聚机理研究进展
2017-06-27黄永旺陈延辉孟雪姣
张 乐,黄永旺,陈延辉,孟雪姣,姜 涛
(天津科技大学 化工与材料学院,天津 300457)
乙烯选择性齐聚机理研究进展
张 乐,黄永旺,陈延辉,孟雪姣,姜 涛
(天津科技大学 化工与材料学院,天津 300457)
乙烯选择性齐聚是生产高级线性α-烯烃的主要方法,具有原子经济性,符合绿色化工的发展趋势,乙烯选择性齐聚的核心在于催化剂,而催化机理研究是开发高催化活性、高选择性乙烯齐聚催化剂的关键。详细阐述了乙烯选择性齐聚反应机理方面研究的最新进展、有效的研究方法及其在乙烯选择性齐聚机理研究中的应用。提出密度泛函理论计算方法、X射线吸收精细结构(XAFS)和电子顺磁共振(EPR)等分析方法的熟练与灵活应用,可为乙烯选择性齐聚反应机理的研究提供很好的帮助,也是该领域发展的一个重要方向。
乙烯选择性齐聚;机理;电子顺磁共振;密度泛函理论;X射线吸收精细结构
高级线性α-烯烃是一类重要的有机化工原料,在聚乙烯共聚单体、表面活性剂、润滑油和油品添加剂等领域有着广泛的应用[1]。随着全球经济的发展和对高性能聚乙烯需求的增加,1-己烯和1-辛烯的需求量以年均5.4%以上的速率增长。乙烯选择性齐聚生产1-己烯、1-辛烯和1-癸烯等高级线性α-烯烃具有工艺流程简单、设备投资少和原子利用率高等优点,是该领域内研究的热点。
近年来有关乙烯选择性齐聚的研究主要集中在新型过渡金属络合物催化剂的开发,所用的金属主要是铬、钛等,研究发现配体的结构对过渡金属络合物乙烯选择性齐聚催化剂的性能有着重要影响[2-28]。目前,对乙烯选择性二聚、三聚和四聚的催化剂研究已有很多文献评述[1,29-35],但由于催化过程中活性中间体的不稳定性及其进行提纯和表征的手段十分有限,对乙烯选择性齐聚反应机理方面的研究较少,而充分认识乙烯选择性齐聚反应机理是开发高催化活性、高选择性乙烯齐聚催化剂的关键。
本文主要介绍了近年来乙烯选择性齐聚机理研究的最新进展、有效的研究方法及其在乙烯选择性齐聚机理研究中的应用。
1 乙烯选择性齐聚机理研究新进展
1.1 单金属环三聚机理
Manyik等[36]首先用单金属环机理解释了乙烯选择性三聚反应,后经Briggs[37]修正形成了目前被广泛接受的乙烯选择性三聚机理,如图1a所示。在该反应路径中,首先两分子的乙烯配位到铬中心,通过氧化加成生成铬金属环戊烷,然后第三个乙烯分子配位到铬中心上,并插入到铬环戊烷中生成铬环庚烷,该七元环通过β-氢转移和还原消除释放一分子的1-己烯。乙烯催化聚合的选择性主要取决于铬金属环上中间体乙烯插入的速率和其分解速率之间的相对平衡。当铬环戊烷的乙烯插入速率明显大于其分解速率时,其金属环状结构能够进一步插入乙烯,扩张形成铬环庚烷;而当乙烯插入铬环庚烷的速率明显小于其分解速率时,则表现出选择性三聚,生成1-己烯。
1.2 单金属环四聚机理
乙烯选择性四聚催化剂的作用机理一直是研究的热点。Overett等[38]采用氘标记技术对其课题组研发的乙烯选择性四聚催化剂的催化机理进行了研究,他们认为乙烯选择性四聚也是通过类似于上述金属环三聚机理实现的。首先,金属铬络合物与两分子的乙烯配位,经氧化偶联反应生成铬环戊烷;然后又一分子的乙烯与铬配位并插入到铬环戊烷中生成铬环庚烷;接着重复上一过程生成铬环壬烷;最后通过还原消除得到1-辛烯。其中,铬环庚烷发生歧化反应后,再通过还原消除生成甲基环戊烷和亚甲基环戊烷。Cloete等[39]的研究结果也与该机理相符。但Tomov等[40]研究发现,如果铬七元环能够扩张生成铬九元环,那么后者仍可进一步扩张生成铬十一元环,按照该反应机理,在聚合过程中不可能高选择性的生成1-辛烯,显然,该机理无法很好的解释这一问题。
1.3 双金属环四聚机理
Peitz等[41]提出了双金属环四聚机理(图1b),用于解释1.2节所述问题。他们认为乙烯选择性四聚的活性中心是两个不相连却又彼此靠近的低价铬原子,每个铬原子都可以独立的生成铬环戊烷,这两个铬环戊烷共同进行还原消除后生成1-辛烯。Licciulli等[42]的研究结果也支持了这一机理。
1.4 单、双配位机理
Britovsek等[43]认为,按照1.3节所述机理,1-己烯和1-辛烯的选择性可通过催化剂的物种形式来控制,即单核催化剂可选择性生成1-己烯,双核催化剂可选择性生成1-辛烯。通过改变铬的浓度就可对乙烯齐聚选择性进行调节。但他们在实验过程中发现,Cr-PNP催化体系的1-己烯/1-辛烯选择性并不受铬浓度的影响。因此他们提出了单、双配位机理(图1c)。该机理与单金属环三聚机理类似,首先生成铬环戊烷,其区别在于:铬环戊烷既可与一个分子的乙烯配位,也可与两个分子的乙烯配位。前者进一步进行乙烯插入生成铬环庚烷,发生还原消除得到1-己烯,即选择性三聚;后者则是铬环戊烷同时与两分子的乙烯配位,然后一分子的乙烯插入生成乙烯基铬环庚烷,接着生成1-辛烯,即选择性四聚。1-己烯和1-辛烯的选择性取决于与单、双乙烯配位的铬环戊烷的形成能力。

图1 乙烯选择性齐聚机理[37,41,43]Fig.1 Mechanism of ethylene selective oligomerization[37,41,43].M:metal ion. a Mononuclear metallacycles mechanism;b Binuclear metallacycles mechanism;c Single and double coordination mechanism
2 乙烯选择性齐聚催化机理的研究方法
2.1 密度泛函理论方法
科研工作者为解释乙烯选择性齐聚过程,先后提出了单金属环机理、双金属环机理及单、双配位机理。但目前乙烯选择性齐聚的机理仍没有定论,其仍是该领域研究的重点和热点问题。密度泛函理论(DFT)方法不但能在电子尺度上研究体系中催化剂的激发态、过渡态,还可对催化机理进行模拟计算,从而很好地预测激发态和过渡态的几何构型、理解催化反应机理等,因此,在乙烯选择性齐聚机理研究领域得到了广泛的应用。Bruin等[44]采用DFT模拟对[η5-C5H4CMe2C6H5]TiCl3/MAO(MAO为甲基铝氧烷)三聚催化体系的催化机理进行了研究,发现七元金属环开环生成1-己烯的Gibbs自由能比第四个乙烯配位的Gibbs自由能低,二者分别为18. 4 kcal/mol和18.5 kcal/mol,因此,更趋向于选择性生成1-己烯。Bhaduri等[45]也利用DFT对乙烯三聚铬催化剂的催化机理进行了研究。由其相对能量分布曲线(图2)可知,对于Cossee机理,中性中间体和阳离子模型系统反应路径的速率控制步骤均为铬环戊烷经β-氢转移生成丁烯基铬中间体的步骤,其活化能垒分别为29.33 kcal/mol和32.53 kcal/mol,说明中性中间体活性物种的三聚机理更趋向于Cossee机理。对于金属环状机理,中性中间体作为起始物显然不可行,而Cr(Ⅱ)-Cr(Ⅳ)阳离子中间体活性物种的三聚机理更趋向于金属环机理。

图2 中性活性催化剂(N0和C0)Cossee型反应路径和阳离子活性催化剂(M0)中间体金属环型反应路径相对能量分布曲线[45]Fig.2 Relative energy profile for Cossee type reaction pathway of neutral(N0and C0) and cationic(M0) active catalyst[45]. NTS1-5,N0-6:the intermediates for Cossee type reaction pathway from N0[Cr(Ⅱ)(C2H4)2Cl2];C0-6,CTS1-5:the intermediates for Cossee type reaction pathway from C0[Cr(Ⅱ)(C2H4)2Cl]+;MTS1-3,M1-4:the intermediates for Cossee type reaction pathway from M0C0[Cr(Ⅱ)(C2H4)2Cl]+.
乙烯选择性齐聚过程中的活性物种是其机理研究的核心问题,对于该活性物种的研究内容主要涉及催化聚合过程中活性中间体的结构及活性金属中心的价态等研究。Netalkar等[46]采用DFT方法对一系列双-α-二亚胺类化合物与Pb(Ⅱ)形成的难以结晶的配合物结构进行了研究,发现氯原子对催化剂的结构及电子分布具有重要影响,对所研究的三种配合物,其氯原子上的未成对电子对HOMO分子轨道的贡献最大,除Pb(Ⅱ)和两个氯离子外,其他一些原子 都对LUMO分子轨道有所贡献。Yang等[47]对Al/吡咯基铬系催化剂进行研究也发现了氯原子在催化乙烯齐聚中的作用,认为氯原子的半稳定性行为是选择性生成1-己烯 的关键因素。大量实验研究发现催化剂的空间几何结构,特别是取代基团的电子效应及空间位阻效应对乙烯齐聚反应的活性和选择性有至关重要的影响。Klemps等[48]对决定PCNCP系列催化剂催化乙烯选择性齐聚的关键步骤进行了计算,研究了P原子上空间位阻与齐聚产物分布之间的关系,发现P原子上取代基(乙基和环己基)的电子效应较小,但大位阻基团不利于CrⅠ-(1-己烯)环状化合物的形成(图3)。Cloete等[39]对N原子上不同取代基的PNP配体及其金属络合物的空间参数(N-sub)进行计算比较,发现N-sub增大有利于1-己烯的生成;而N-sub减小有利于1-辛烯和环状C6副产物的生成。Kim等[20]对DPPDME/Cr手性催化体系催化乙烯四聚过程中环扩张阶段的中间体(图4A和B)进行了DFT计算,发现SS型或RR型旋光异构体比内消旋体铬环庚烷的 P—Cr键长更短,结构更稳定,更有利于七元环进一步扩张为九元环,从而选择性生成1-辛烯。
金属中心价态的研究对乙烯选择性齐聚机理及助催化剂的作用机理研究有重要的指导作用。催化剂中铬阳离子氧化态的分析检测已经成为该领域研究的重点。不同氧化态催化剂活性物种的分离对于合成过程来说具有难度,而DFT方法是很好的研究活性物种金属氧化态的理论方法。Yang等[47]通过DFT方法对Al/吡咯基铬催化剂选择性三聚的Cr(Ⅰ)/Cr(Ⅲ)和Cr(Ⅱ)/Cr(Ⅳ)型反应历程的有效活化能进行了计算(图5),分别为19.0 kcal/ mol和31.4 kcal/mol,表明该催化体系更有可能采取Cr(Ⅰ)/Cr(Ⅲ)型反应历程。Albahily等[49-50]报道了一种用于乙烯选择性齐聚的自活化铬基催化剂(图4C),表面上看,三个铬原子中有一个二价和两个一价铬原子,但DFT计算结果表明:两个与丁二烯基配位的铬原子上的一对电子转移至与其相连的丁二烯基的π键系统内,从而三个铬原子均为二价。
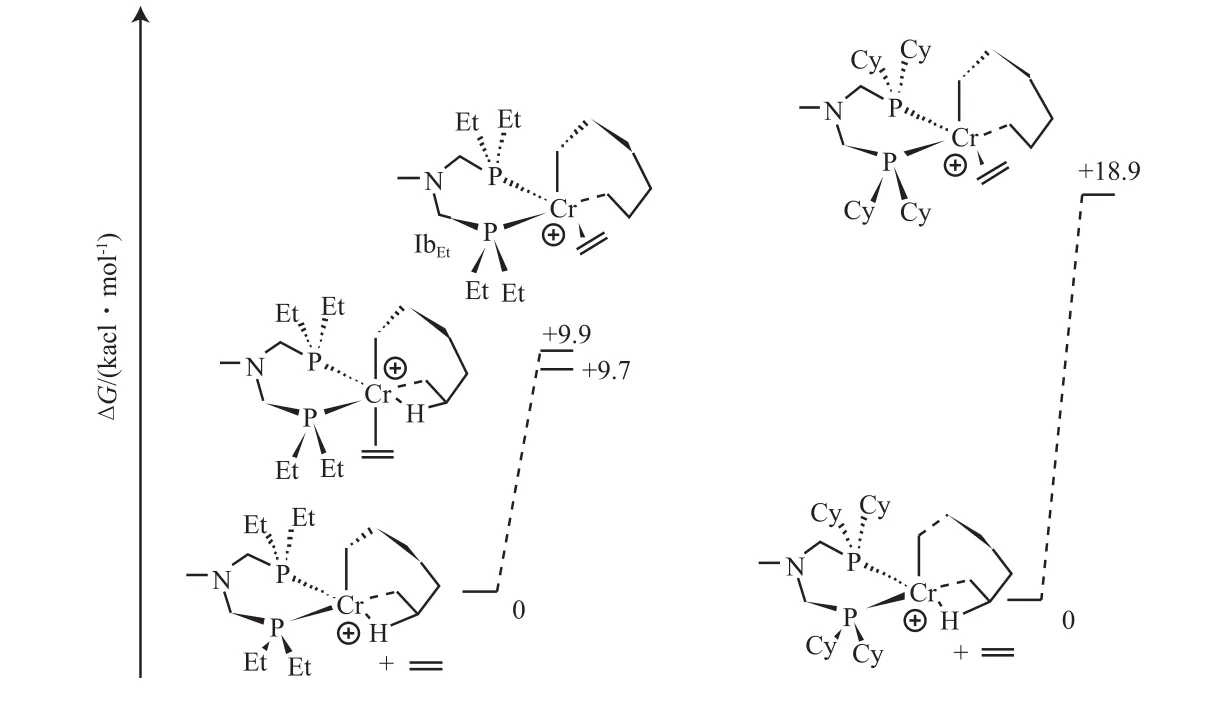
图3 不同膦基团的PCNCP/Cr 环庚烷与乙烯键合的活化自由能[48]Fig.3 Activative energy of the bonding of ethylene to PCNCP/Cr cyclopentane with different substituent group on phosphorus[48].

图4 PNP/Cr(A)和DPPDME/Cr(B)催化体系的金属铬环庚烷结构以及自活化乙烯齐聚催化剂结构(C)[20,49-50]Fig.4 Structures of chromiumcycloheptanes in PNP/Cr(A) and DPPDME/Cr(B) catalytic system and the structure of self-activated catalyst for ethylene oligomerization(C)[20,49-50].

图5 DFT计算的Cr(Ⅰ)/Cr(Ⅲ)(A)和Cr(Ⅱ)/Cr(Ⅳ)(B) 型反应历程的自由能级图[47]Fig.5 Free energy profiles of Cr(Ⅰ)/Cr(Ⅲ)(A) and Cr(Ⅱ)/Cr(Ⅳ)(B) reaction routes calculated by density functional theory[47].
除氧化态金属外,催化过程中金属中心自旋态的转变对催化反应活性也有重要影响,因此也引起了科研工作者们的关注。由于乙烯选择性齐聚的活性物种难以捕获,因此对自旋态的实验研究鲜有报道。DFT方法在活性物种自旋态的研究方面也取得了一定的成果。Klemps等[48,51]的研究发现,在不存在自旋交叉的催化系统中,Cr(Ⅰ)/Cr(Ⅲ)氧化耦合以四旋态较为稳定;而Janes等[52]研究发现Cr(Ⅱ)/Cr(Ⅳ)氧化耦合则以三旋态最为稳定。Yang等[53-54]的理论研究也揭示了两种自旋态在乙烯二聚和三聚中的可能性,且自旋加速促进了反应活性的提高。
DFT方法除了在机理研究方面被广泛应用之外,还被Gong等[55]用于[CrCl3(PNPOMe)]催化乙烯/己烯三聚产物的分布研究中,他们以金属环机理为基础,对不同十碳烯异构体生成路径的表面自由能进行计算,合理解释了实验数据十碳烯异构体的分布情况。
2.2 光谱研究方法
由于大多数铬配合物的顺磁特性和MAO的复杂特性,常用于分析有机及金属有机化合物的核磁共振光谱分析法难以对铬催化剂及其反应过程中的中间体进行结构表征,阻碍了乙烯选择性齐聚反应机理的研究进展。X射线吸收精细结构(XAFS)光谱和电子顺磁共振(EPR)光谱则可以绕过被检测物质的顺磁性壁垒,对顺磁性铬化合物的局部结构和铬金属的中心价态进行表征。
XAFS光谱的基本原理是通过增加入射X光子的能量,使光电子波长逐渐变短,出射光与邻近原子的散射光电子波之间的干涉,使得吸收曲线产生震荡,其细节部分包含吸收原子周围及邻近原子短程结构信息,既可用于非晶、液态、熔态、金属蛋白、晶体中的杂质原子,也可用于催化剂活性中心的结构研究。Bartlett等[56]以Mo类似物代替用于乙烯选择性三聚的Cr催化剂,在截流系统中用时间分辨Mo原子K边缘扩展XAFS光谱,探讨了其在MAO作用下的活化过程,发现Mo化合物在过量的AlMe3作用下失去卤素原子,形成[Mo(CH3)3(L)],且在n(Mo)∶n(AlMe3)= 1∶20条件下,系统中既没有Mo—Mo键合,也没有卤素桥联的Mo二聚体存在。该结论在一定程度上支持了乙烯选择性齐聚的单核机理。Nenu等[57]对二氧化硅负载的Phillips型乙烯三聚催化系统Cr/TAC/CH2Cl2(TAC:1,3,5-三苄基六氢-1,3,5-三嗪)组装过程中的Cr原子K/L2,3边缘吸收光谱进行了研究。结果表明:Cr原子与TAC和含氯配体以及二氧化硅的晶格相连,形成了新的单活性位。在此组装过程中,Cr的价态先由正六价转变为正二价,再转变为正三价,且Cr配合物的八面体几何构型得以保持,但有明显的扭曲变化。在此基础上,Bartlett等[58]采用冷冻淬灭技术和Cr的K边缘扩展XAFS光谱对CrCl3(THF)3/N(iPr)(PPh2)2(THF为四氢呋喃)催化剂进行了研究,发现该催化剂在液氮冷冻下与过量的AlMe3作用时,首先生成单甲基化的CrClMe(ClAlCl3){PPh2N(R)PPh2}(THF)(R为甲基)物种,随后逐渐转化为高自旋态的CrCl2{PPh2N(R)PPh2}二价铬物种;而在常温下,则生成CrClMe{PPh2N(R)PPh2}。此结果揭示了烷基铝对Cr系四聚催化剂的活化过程。
EPR光谱对处于催化循环中Cr的某些氧化态是活跃的,而对于某些氧化态是无声的,因此,其可以同时监测催化系统中的活性物种及亚活性物种。EPR光谱技术很早就被用于鉴定顺磁性金属化合物及监测它们的氧化或还原反应过程[59-60],但在乙烯选择性齐聚领域,直到2008年Brückner等[61]才首次报道了利用原位EPR光谱研究乙烯齐聚催化剂的演变历程。该报道对Cr(acac)3/ PNP(acac为乙酰丙酮阴离子)原位系统和分离纯化的[(PNP)CrCl2(μ-Cl)]2催化系统进行了分析比较。通过对Cr(acac)3/PNP/MMAO(MMAO为改性MAO)原位系统在高温高压下与乙烯相互作用的EPR光谱研究(其EPR光谱见图6A和B),发现了Cr+与PNP配体的络合物,而没有发现无配体配位的Cr+物种的存在。研究还发现,上述Cr+与PNP配体的络合物的含量在总铬中的比例很小,推测Cr+/PNP化合物可能是一种短暂的存在形式;其主要以另外一种处于亚活性状态的抗铁磁性的Cr+二聚体形式存在,但该二聚体无法用EPR光谱监测。从[(PNP)CrCl2(μ-Cl)]2/MMAO催化系统在甲苯中的各向同性EPR光谱中发现了一个尖锐的多重超精细结构的EPR信号谱图(图6C和D),通过对各向同性值giso和Aiso及电子自旋(s = 1/2)与53Cr同位素核自旋的超精细耦合常数比较,证明该多重谱线归属于具有三明治结构的[Cr(η6-CH3C6H5)2]+阳离子。该物种的形成抑制了抗铁磁性Cr+二聚体的形成,从而导致活性低下,说明对于该系统,甲苯不是理想的反应溶剂。
在此基础上,Skobelev等[62]也采用EPR光谱研究了Phillips乙烯三聚催化剂和催化剂不同组成对Cr(Ⅲ)和Cr(Ⅰ)的影响。通过对Cr(acac)3/ AlEt3和Cr(acac)3/parole/AlEt3催化系统EPR光谱的比较发现,高活性催化系统具有高浓度的单核Cr(Ⅰ)物种(图7)。因此,反应混合物中单核Cr(Ⅰ)的存在与三聚活性有关。在对n(Cr(EH)3)∶n(HPyr)∶n(AlEt3)= 1∶3∶30(EH为异辛酸阴离子)和n(Cr(EH)3)∶n(AlEt3)= 1∶30,n(Cr(acac)3)∶n(AlEt2Cl)= 1∶20的研究中也得到了类似的数据。所有数据均指明Cr(Ⅰ)物种参与了乙烯三聚的催化反应。因此推断Cr(Ⅰ)-Cr(Ⅲ)机理是最有可能的Phillips催化剂催化反应历程。
EPR光谱是研究顺磁性有机金属催化剂及其中间体的有力工具,其在乙烯齐聚催化机理的研究中也得到了较为广泛的应用。但EPR光谱对于某些价态的金属化合物表现为无声状态,在其应用中存在着一定的缺陷。多种光谱技术联用成为催化机理研究领域的研究热点,如X波段连续波电子-原子核双共振波谱(CW-EPR)与电子-核双共振(ENDOR)结合[63];EPR与UV-Vis、EXAFS和XANES结合[64-65]等。

图6 Cr(acac)3/PNP/MMAO的原位EPR光谱(A和B)和[(PNP)CrCl2(μ-Cl)]2在甲苯溶液中的EPR光谱(C和D)[61]Fig.6 In situ EPR spectrum of Cr(acac)3/PNP/MMAO and[(PNP)CrCl2(μ-Cl)]2in toluene[61].B:the magnetic filed vector;giso:the isotropic g tensor;Aiso:the isotropic hyperfine coupling tensor.(A) Duiring isothermal treatment at 333 K with 1 MPa of ethylene;(B) After the reaction at room temperature(black line) and 77 K(gray line);(C) Measured at 293 K after 5 min(solid line) and 15 min(dashed line) contact with MMAO;(D) Second derivative of the spectrum after 15 min

图7 不同条件处理的Cr(acac)3/AlEt3试样(A)和Cr(acac)3/HPyr/AlEt3(B)的EPR光谱Fig.7 EPR spectra of the samples of Cr(acac)3/AlEt3(A) and Cr(acac)3/HPyr/AlEt3(B) after various treatment.(a)-(c) The EPR spectra of the samples of A storing at 20 ℃ for 1,6,15 min,respectively;(d) The one of sample A stored at 65 ℃ for 1 min;(e)-(g) The EPR spectra of the samples of B storing at 20 ℃ for 1,4,28 min,respectively
3 结语
催化机理的研究一直是乙烯选择性齐聚领域的研究重点和难点。目前,比较公认的乙烯选择性齐聚机理是单金属环机理和双金属环机理。前者最先用来解释选择性三聚,后来进一步应用于选择性四聚。但大量实验数据表明,选择性四聚反应产物中,C10以上线性α-烯烃产物的选择性很低,这一点很难用金属环状机理来解释。双金属协同机理则很好地解释了这一现象。但对于双金属协同机理的实验证明还相当有限,有待于大量的实验数据予以证明。此外,按照以上两种机理,乙烯选择性五聚、六聚几乎是不可能实现的。然而,最近提出的单、双配位机理则从金属配位化学的角度,为乙烯选择性齐聚的机理研究提出了新的方向,认为乙烯选择性齐聚的关键在于三个或四个乙烯单元与金属中心配位化合物能否稳定存在。从该机理出发,如果能够通过配体和金属的合理设计与搭配,使得金属中心能够同时配位五个甚至更多的乙烯单元,那么很有可能在乙烯选择性五聚、六聚等方面实现突破。
催化剂中间体的分离提纯与表征一直是制约催化机理研究发展的瓶颈问题。DFT计算方法不仅能够对中间体结构进行优化,得到空间结构参数,弥补无法得到单晶结构信息的不足,还能够对反应过程的活化能进行计算,从反应热力学角度为机理研究提供理论基础。而XAFS和EPR等光谱分析方法则可绕过某些活性中心的顺磁性壁垒,对其金属价态、自旋态及其结构进行表征。以上几种方法,在催化领域早有应用,而在乙烯选择性齐聚方面,其应用还不是十分广泛。这些技术方法的熟练与灵活应用,能为其反应机理的研究提供很好的帮助,也是该领域发展的一个重要方向。
[1] Agapie T. Selective ethylene oligomer ization:Recent advances in chromium catalysis and mechanistic investigations[J].Coord Chem Rev,2011,255(7):861-880.
[2] Weng Z,Teo S,Hor T A. Chromium (Ⅲ) catalysed ethylene tetramerization promoted by bis(phosphino) amines with an N-functionalized pendant[J].Dalton Trans,2007,44(32):3493-3498.
[3] Stennett T E,Hey T W,Ball L T,et al. N,N-diphospholylamines—A new family of ligands for highly active,chromium-based,selective ethene oligomerisation catalysts[J].C hem Cat Chem,2013,5(10):2946-2954.
[4] Sa S,Lee Sung Min,Kim Sang Youl. Chromium-based ethylene tetramerization with diphosphinoamines bearing pendent amine donors[J].J Mol Catal A:Chem,2013,378(11):17-21.
[5] Overett M J,Blann K,Bollmann A,et al. Ethylene trimerisation and tetramerisation catalysts with polar-substituted diphosphinoamine ligands[J].Chem Commun,2005,5(5):622-624.
[6] Mao Guoliang,Ning Yingnan,Hu Wenbo,et al. Synthesis of a novel triple-site diphosphinoamine(PNP) ligand and its applications in ethylene tetramerization[J].Chin Sci Bull,2008,53(22):3511-3515.
[7] Kuhlmann S,Blann K,Bollmann A,et al. N-substituted diphosphinoamines:Toward rational ligand design for the effi-cient tetramerization of ethylene[J].J Catal,2007,245(2):279-284.
[8] Killian E,Blann K,Bollmann A,et al. The use of bis (diphenylphosphino) amines with N-aryl functionalities in selective ethylene tri-and tetramerization[J].J Mol Catal A:Chem,2007,270(1):214-218.
[9] Jiang Tao,Zhang Sai,Jiang Xing ling,et al. The effect of N-aryl bisphosphineamine ligands on the selective ethylene tetramerization[J].J Mol Catal A:Chem,2008,279(1):90-93.
[10] Jiang Tao,Tao Yiqing,Gao Xianglu,et al. Ethylene tetramerization with a highly active and long-lifetime trinuclear diphenylphosphinoamine/Cr(Ⅲ)/MAO catalyst[J].Chin Sci Bull,2012,57(13):1510-1515.
[11] Jiang Tao,Chen Hongxia,Ning Yingnan,et al. Highly selective diphoshinoamine/Cr(Ⅲ)catalysts for ethylene tetramerization[J].Chin Chem Lett,2006,17(3):358.
[12] Jiang Tao,Chen Hongxia,Ning Yingnan,et al. Preparation of 1-octene by ethylene tetramerization with high selectivity[J].Chin Sci Bull,2006,51(5):521-523.
[13] Elowe P R,Mc Cann C,Pringle P G,et al. Nitrogen-linked diphosphine ligands with ethers attached to nitrogen for chromium-catalyzed ethylene tri-and tetram erizations[J].Organometallics,2006,25(22):5255-5260.
[14] Carter A,Cohen S A,Cooley N A,et al. High activity ethylene trimerisation catalysts based on diphosphine ligands[J]. Chem C ommun,2002, (8):858-859.
[15] Blann K,Bollmann A,Bod H D,et al. Ethylene tetramerisation:Subtle effects exhibited by N-substituted diphosphinoamine ligands[J].J Catal,2007,249(2):244-249.
[16] Bollmann A,Blann K,Dixon J T,et al. Ethylene tetra merization:A new route to produce 1-octene in exceptionally high selectivities[J].J Am Chem Soc,2004,126(45):14712-14713.
[17] Shai kh Y,Albahily K,Sutcliffe M,et al. A highly selective ethylene tetramerization catalyst[J].Angew Chem,Int Ed,2012,51(6):1366-1369.
[18] Dulai A,De Bod H,Hanton M J,et al. C- substituted bis(diphenylphosphino) methane-type ligands fo r chromium-catalyzed selective ethylene oligomerization reactions[J]. Organometallics,2009,28(15):4613-4616.
[19] Han T-K,Kang S-O,Kim S-K. Highly active and selective ethylene oligomerization catalyst and method of preparing hexene or octene using the sam:US8829218[P].2014-09-09.
[20] Kim Sung-Kwan,Kim Tae-Jin,Chung Jae-Ho,et al. Bimetallic ethylene tetramerization catalysts derived from chiral dppdme ligands:Syntheses,structural characterizations,and catalytic performance of[(dppdme) CrCl3]2(dppdme = S,S- and R,R-Chirap hos and Meso-Achiraphos)[J].Organometallics,2010,29(22): 5805-5811.
[21] Overett M J,Blann K,Bollmann A,et al. Carbon-bridged diphosphine ligands for chromium-catalysed ethylene t etramerisation and trimerisation reactions[J].J Mol Cat al A:Chem,2008,283(1):114-119.
[22] Zhang Jun,Wang Xiao,Zhang Xuejun,et al. Switchable ethylene tri-/tetramerization with high activity:Subtle effe ct presented by backbone-substituent of carbon-bridged diphosphine ligands[J].ACS Catal,2013,3(10):2311-2317.
[23] Dulai A,McMullin C L,Tenz a K,et al. N,N′-bis (diphenylphosphino) diaminophenylphosphine ligands for chromiumcatalyzed selective ethylene oligomerization reactions[J]. Organometallics,2011,30(5):935-941.
[24] Härzschel S,Kühn F E,Wöhl A,et al. Comparative study of new chromium-based catalysts for the selective tri- and tetramerization of ethylene[J].Catal Sci Technol,2015,5(3):1678-1682.
[25] Shaikh Y,Gurnham J,Albahily K,et al. Aminophosphinebased chromium catalysts for selective ethylene tetramerization[J].Organometallics,2012,31(21):7427-7433.
[26] Zhou Yusheng,Wu Hongfei,Xu Sh eng,et al. Highly active chromium-based selective ethylen e tri-/tetramerization catalysts supported by PNPO phosphazane ligands[J].Dalton Trans,2015,44(20):9545-9550.
[27] McGuinness D S,Wasserscheid P,Keim W,et al. First Cr(Ⅲ)-SNS complexes and their use as highly efficient catalysts for the trimerization of ethylene to 1-hexene[J].J Am Chem Soc,2003,125(18):5272-5273.
[28] Yoshida Toru,Yamamoto Toshihide,Oka da Hisanori,et al. Catalyst for trimerization of ethylene and process for trimerizing ethylene using the catalyst:US6900152[P].2005-03-31.
[29] 董博,孙月明,王媚,等. 乙烯四聚合成1-辛烯研究新进展 [J].高分子通报,2013(12):38-43.
[30] Dixon J T,Green M J,Hess F M,et al. Advances in selective ethylene trimerisation—A critical overview[J].J Organomet Chem,2004,689(23):3641-3668.
[31] Wass D F. Chromium-catalysed ethene trimerisation and tetramer isation—Breaking the rules in olefin oligomerisation[J]. Dalton Trans,2007,62(8):816-819.
[32] Belov G. Selective dimerization,oligomerization,homopolymerizat ion and copolymerization of olefins with complex organometallic catalysts[J].Russ J Appl Chem,2008,81(9):1655-1666.
[33] van Leeuwen P W,Clément N D,Tschan M J-L. New processes for the selective production of 1-octene[J].Coord Chem Rev,2011,255(13):1499-1517.
[34] Belov G. Tetramerization of ethylene to octene-1(A review)[J].Pet Chem,2012,52(3):139-154.
[35] McGuinness D S. Olefin oligomerization via metallacycles:Dimeriza tion,trimerization,tetramerization,and beyond[J].Chem Rev,2010,111(3):2321-2341.
[36] Manyik R,Walker W,Wilson T. A soluble chromium-based catalyst for ethylene trimerization and polymerization[J].J Catal,1977,47(2):197-209.
[37] Briggs J R. The selective trimerization of ethylene to hex1-ene[J].J Ch em Soc Chem Commun,1989 ,11 (11):674-675.
[38] Overett M J,Blann K,Bollmann A,et al. Mechanistic investigatio ns of the ethylene tetramerisation reaction[J].J Am Chem Soc,2005,127(30):10723-10730.
[39] Cloete N,Visser H G,Engelbrecht I,et al. Ethylene tri-and tetra merization:A steric parameter selectivity switch from X-ray crystallography and computational analysis[J].Inorg Chem,2013,52(5):2268-2270.
[40] Tomov A K,Chirinos J J,Jones D J,et al. Experimental evidence f or large ring metallacycle intermediates in polyethylene chain growth using homogeneous chromium catalysts[J]. J Am Chem Soc,2005,127(29):10166-10167.
[41] Peitz S,Aluri B R,Peulecke N,et al. An alternative mechanistic concept for homogeneous selective ethylene oligomerization of chromium-based catalysts:Binuclear metallacycles as a reason for 1-octene selectivity[J].Chem,2010,16(26):7670-7676.
[42] Licciulli S,Thapa I,Albahily K,et al. Towards selective ethylen e tetr amerization[J].Angew Chem,Int Ed,2010,49(48):9225-9228.
[43] Britovsek G J,McGuinness D S,Wierenga T S,et al. Single-and doub le-coordination mechanism in ethylene tri- and tetramerization with Cr/PNP catalysts[J].ACS Catal,2015,5(7):4152-4166.
[44] Bruin T J M D,M agna L,Raybaud P,et al. Hemilabile ligand induce d selectivity:A DFT study on ethylene trimerization catalyzed by titanium complexes[J].Organometallics,2003,22(17):3404-3413.
[45] Bhaduri S,Mukhopadhyay S,Kulkarni S A. Density functional studie s on chromium catalyzed ethylene trimerization[J].J Organomet Chem,2009,694(9):1297-1307.
[46] Netalkar S P,Budagumpi S,Abdallah H H,et al. Sterically modulate d binuclear bis-α-diimine Pd (Ⅱ) complexes:Synthesis,characterization,DFT studies and catalytic behavior towards ethylene oligomerization[J].J Mol Struct,2014,1075:559-565.
[47] Yang Yun,Liu Zhen,Cheng Ruihua,et al. Mechanistic DFT study on eth ylene trimerization of chromium catalysts supported by a versatile pyrrole ligand system[J].Organometallics,2014,33(10):2599-2607.
[48] Klemps C,Payet E,Magna L,et al. PCNCP Ligands in the chromium-cata lyzed oligomerization of ethylene:Tri-ve rsus tetramerization[J].Chem Eur J,2009,15(33):8259-8268.
[49] Albahily K,Fomitcheva V,Gambarotta S,et al. Preparation and charac terization of a reduced chromium complex via vinyl oxidative coupling:Formation of a self-activating catalyst for selective ethylene trimerization[J].J Am Chem Soc,2011,133(16):6380-6387.
[50] Albahily K,Shaikh Y,Sebastiao E,et al. Vinyl oxidative coupling as a synthetic route to catalytically active monovalent chromium[J].J Am Chem Soc,2011,133(16):6388-6395.
[51] Budzelaar P H. Ethene trimerization at Cr(Ⅰ)/Cr(Ⅲ)—A density function al theory(DFT) study[J].Can J Chem,2009,87(7):832-837.
[52] van Rensburg W J,Grové C,Steynberg J P,et al. A DFT study toward the mechanism of chromium-catalyzed ethylene trimerization[J].Organometallics,2004,23(6):1207-1222.
[53] Yang Yun,Liu Zhen,Zhong Lei,et al. Spin surface crossing between chrom ium (Ⅰ)/sextet and chromium (Ⅲ)/quartet without depro tonation in SNS-Cr mediated ethylene trimerization[J].Organometallics,2011,30(19):5297-5302.
[54] Liu Zhen,Zhong Lei,Yang Yun,et al. DFT and CASPT2 study on the mechanism of ethylene dimerization over Cr (Ⅱ)OH+ cation[J]. J Phys Chem A,2011,115(28):8131-8141.
[55] Gong Minglan,Liu Zhen,Li Yuanhui,et al. Selective cooligomerization of e thylene and 1-hexene by chromium-PNP catalysts:A DFT st udy[J].Organometallics,2016,35(7):972-981.
[56] Bartlett S A,Wells P P,Nachtegaal M,et al. Insights in the mechanism of s elective olefin oligomerisation catalysis using stopped-flow freeze-quench techniques:A Mo K-edge QEXAFS study[J].J Catal,2011,284(2):247-258.
[57] Nenu C N,van Lingen J N,de Groot F M,et al. Controlled assembly of a hete rogeneous single-site ethylene trimerization catalyst as probed by X-Ray absorption spectroscopy[J]. Chem—Eur J,2006,12(18):4756-4763.
[58] Bartlett S A,Moulin J,Tromp M,et al. Activation of[CrCl3{PPh2N(iPr) PPh2}]for the selective oligomerisation of ethene:A Cr K-edge XAFS study[J].Catal Sci T echnol,2016,6:6237-6246.
[59] Doorslaer S V,Caretti I,Fallis I A,et al. The power of electron paramagne tic resonance to study asymmetric homogeneous catalysts based on transition-metal complexes[J].Coord Chem Rev,2009,253(15):2116-2130.
[60] Doorslaer S V,Murphy D M. EPR spectroscopy in catalysis[M].Berlin Heidelbe rg:Springer,2011:1-39.
[61] Brückner A,Jabor J K,Mcconnell A E C,et al. Monitoring structure and valen ce st a te of chromium sites during catalyst formation and ethylene oligomerization by in situ EPR spectroscopy[J].Organometallics,2008,27(15):3849-3856.
[62] Skobelev I Y,Panchenko V N,Lyakin O Y,et al. In situ EPR monitoring of chro mium species formed during Cr-pyrrolyl ethylene trimerization catalyst formation[J].Organometallics, 2010,29(13):2943-2950.
[63] Mcdyre L E,Hamilton T,Murphy D M,et al. A cw EPR and ENDOR investigation on a ser ies of Cr (I) carbonyl complexes with relevance to alkene oligomerization catalysis:[Cr (CO)4L]+(L=Ph2PN(R)PPh2,Ph2P(R)PPh2)[J]. Dalton Trans,2010,39(39):7792-7799.
[64] Moulin J O,Evans J,McGuinness D S,et al. Probing the effects of ligand stru cture on activity and selectivity of Cr (Ⅲ)complexes for ethylene oligomerisation and polymerisation[J].Dalton Trans,2008,252(9):1177-1185.
[65] Rabeah J,Bauer M,Baumann W,et al. Formation,operation and deactivation of Cr catalysts in ethylene tetramerization directly assessed by operando EPR and XAS[J].ACS Catal,2012,3(1):95-10.
(编辑 平春霞)
Advances in mechanistic research of ethylene selective oligomerization
Zhang Le,Huang Yongwang,Chen Yanhui,Meng Xuejiao,Jiang Tao
(College of Chemical Engineering and Material Science,Tianjin University of Science and Technology,Tianjin 300457,China)
Advanced linear α-olefins are mainly produced by selective oligomerization of ethylene. Benefiting from the atom economy,selective oligomerization is in accord with the development tendency of green chemistry. Catalyst is the core of selective oligomerization and the catalytic mechanism is the key for developing the catalyst wit h high activity and selectivity. Recent development of the mechanism for ethylene oligomerization,efficientive research method and their application are reviewed in detail. The skilled and flexible application of density functional theory and the spectrometry such as X-ray absorption fine structure(XAFS) and electron paramagnetic resonance(EPR) is quite helpful for the ethylene oligomerization mechanism,which is an important research field in the future.
ethylene selective oligomerization;mechanistic;electron paramagnetic resonance;density functional theory;X-ray absorption fine structure
1000-8144(2017)06-0791-10
TQ 316.3
A
10.3969/j.issn.1000-8144.2017.06.023
2016-11-14;[修改稿日期] 2017-01-10。
张乐(1985—),男,河北省邯郸市人,博士生,电话 18617598080,电邮 josephgo@sina.com。联系人:姜涛,电话 022-60602936,电邮 jiangtao@tust.edu.cn。
天津市应用基础与前沿技术研究计划重点项目(16JCZDJC31600);国家自然科学基金委员会-中国石油天然气集团公司石油化工联合基金资助项目(U1162114)。
