大黑毛肩长蝽(半翅目:异翅亚目:地长蝽科)线粒体基因组及地长蝽科系统发育地位探讨
2017-06-05郭鹏磊吴燕卓王艳会吴昊阳李俊兰
郭鹏磊,吴燕卓,王艳会,吴昊阳,李 腾,李俊兰,
卜文俊1,谢 强4*
大黑毛肩长蝽(半翅目:异翅亚目:地长蝽科)线粒体基因组及地长蝽科系统发育地位探讨
郭鹏磊1,吴燕卓1,王艳会1,吴昊阳1,李 腾2,李俊兰3,
卜文俊1,谢 强4*
(1. 南开大学生命科学学院,天津 300071;2. 兰州大学生命科学学院,兰州 730000;3. 内蒙古大学生命科学学院,呼和浩特 010021;4. 生物多样性演化与保护广东普通高校重点实验室,中山大学生物博物馆,中山大学生命科学学院,广州 510275)
为了更加深入地了解地长蝽科的基因组水平特征,测序并分析了大黑毛肩长蝽Neolethaeusassamensis(半翅目:异翅亚目:地长蝽科:毛肩族)的线粒体基因组序列。大黑毛肩长蝽线粒体基因组是双链共价环状DNA分子,长度为17097 bp,编码13个蛋白质编码基因,22个tRNA基因和2个rRNA基因,基因排列方式同果蝇Drosophilayakuba一致。大黑毛肩长蝽线粒体基因组内存在2个大的非编码区。一个是控制区,另一个是位于ND6和CytB之间的串联重复区域,TRR4.4。控制区内包含7类共9个结构显著的区域,如一个茎环结构,3个非串联的重复序列以及其他5个结构区域。TRR4.4长802 bp,包括4个184 bp的重复单元和1个66 bp的部分重复单元。TRR4.4的重复单元与控制区中TRR2.7的重复单元在长度、方向以及核苷酸组成等方面几乎完全一致。22个tRNA全部能够折叠为典型的三叶草二级结构。16S rRNA二级结构包含6个结构域(结构域III在节肢动物中缺失)和44个茎环结构,12S rRNA二级结构包含3个结构域和28个茎环结构。基于蝽次目15个线粒体基因组数据分析得到的系统发育结果,支持地长蝽科位于长蝽总科基部分支的观点。
线粒体基因组;大黑毛肩长蝽;串联重复序列;重组;系统发育
The complete mitochondrial genome ofNeolethaeusassamensis
线粒体是真核细胞中重要的细胞器,拥有独立的遗传物质。在六足类动物中,线粒体基因组(mitochondrial genome)是双链环状DNA分子,大小约14-20 kb。整个线粒体基因组一般由一个控制区(control region,CR)和37个基因组成,其中包括13个蛋白质编码基因(PCGs),22个tRNA基因和2个rRNA基因(Wolstenholme, 1992;Boore, 1999)。控制区由于富含碱基AT,又被称为A+T丰富区(Stormo, 1982)。控制区在线粒体基因组中具有重要的作用:一方面,控制区内含有调控转录和复制起始的信息位点(Wolstenholme, 1992;Zhang and Hewitt, 1997);另一方面,线粒体基因组的大小主要受到控制区内串联重复序列的影响(Zhang and Hewitt, 1997)。线粒体基因组具有重组率低、进化速率快和结构保守等特点,被广泛应用于系统发育学、种群遗传学和生物地理学等研究领域(Sacconeetal., 1999;Cameron, 2014a)。
地长蝽科Rhyparochromidae是半翅目Hemiptera长蝽总科Lygaeoidea中最大的科,目前世界已知约372属1850种(Henry, 2009)。地长蝽科最为显著的特征是腹面第四、五腹节的节间缝通常向前斜伸,但很少到达腹部侧缘。地长蝽科昆虫一般为中小体型,身体呈黑褐色、黄褐色或黑白相间。大部分种类在地表生活,以成熟脱落于地面的种子为食;少数种类藏于果穗中,吸食种子(郑乐怡和邹环光,1981;Schuh and Slater, 1995)。
目前为止,GenBank中共收录了长蝽总科7个线粒体基因组(包括全序列和部分序列)(表1),但是地长蝽科仅报道了地栖族Rhyparochromini中白斑地长蝽Panaorusalbomaculatus的线粒体基因组,与地长蝽科庞大的物种数量严重不符。为了更全面的了解长蝽总科的系统发育关系,需要获取地长蝽科内更多物种的线粒体基因组数据。本研究测序了地长蝽科毛肩族Lethaeini中大黑毛肩长蝽NeolethaeusassamensisDistant, 1901的线粒体基因组,并且分析和描述了大黑毛肩长蝽线粒体基因组相关结构特征,例如碱基组成、密码子使用、tRNA和rRNA的二级结构以及非编码区域等。
1 材料与方法
1.1 标本采集
大黑毛肩长蝽的成虫个体,在2015年8月份采自于云南省瑞丽市珍稀植物园,在野外所有标本存放在无水乙醇中,回到实验室后置于-20℃环境保存。
1.2 DNA提取
取大黑毛肩长蝽的胸部肌肉组织,采用CTAB法(Reinekeetal., 1998)提取全DNA,-20℃环境存放。

表1 本研究中所使用的类群信息
1.3 引物设计、PCR扩增和测序
大黑毛肩长蝽线粒体基因组序列是通过扩增14个具有部分重叠序列的DNA片段得到的。DNA片段:ND2,COI,COIII,ND5,ND4以及16S rDNA-12S rDNA使用的PCR引物参照长蝽总科线粒体基因组的通用引物(Lietal., 2013);DNA片段:ND2-COI,COI-COIII,COIII-ND5,ND5-ND4,ND4-ND6,ND6-CytB,CytB-16S rDNA和12S rDNA-ND2使用的PCR引物是利用相关引物设计软件设计的物种特异性引物(表2)。使用TaKaRa LA DNA聚合酶进行PCR扩增,PCR反应过程:94℃预变性2 min;94℃变性30 s,45℃-55℃退火30 s,72℃延伸1-4 min,共进行34个循环;72℃终延伸10 min。使用1%琼脂糖凝胶电泳检测PCR产物,之后送交生物服务公司(BGI,深圳)进行双向测序。

表2 本研究中使用的PCR引物
1.4 DNA序列拼接、注释及分析
在NCBI中利用Blast功能比对DNA序列,确定是目的基因后,使用BioEdit v.7.0.9.0(Hall, 1999)依据相邻片段间的重叠区域进行DNA序列拼接。
线粒体基因组的注释参考Cameron(2014b)中报道的方法,首先分析注释tRNA基因 和rRNA基因,其次分析蛋白质编码基因的范围,最后探讨控制区的相关结构。
tRNA基因的注释通过2种在线软件,tRNAscan-SE v1.21(http://lowelab.ucsc.edu/tRNAscan-SE/)(Lowe and Eddy, 1997)和MITOS WebServer(http://mitos.bioinf.uni-leipzig.de/index. py/)(Berntetal., 2013)。不能直接查找到的tRNA基因,根据已知半翅目线粒体基因组中相对应的tRNA确定其位置,并通过RNAstructure 5.8(Reuter and Mathews, 2010)推测其二级结构。
rRNA基因通常被注释为相邻两个基因间的区域(Boore, 2001;Cameron, 2014b)。rRNA二级结构的构建主要依据已发表的昆虫线粒体基因组rRNA的二级结构模型,例如黑腹果蝇Drosophilamelanogaster(双翅目:果蝇科)(Cannoneetal., 2002),Aradacanthiaheissi(半翅目:异翅亚目:扁蝽科)(Shietal., 2012),豆突眼长蝽Chauliopsfallax(半翅目:异翅亚目:束长蝽科)(Lietal., 2013),异色巨蝽Eusthenescupreus(半翅目:异翅亚目:荔蝽科)(Songetal., 2013)和亚姬缘蝽Corizustetraspilus(半翅目:异翅亚目:姬缘蝽科)(Yuanetal., 2015)。其中茎环结构的命名采用Cameron and Whiting(2008)和Gillespie等(2006)的命名规则。使用RNAstructure 5.8(Reuter and Mathews, 2010)预测缺乏同源结构的片段。
利用NCBI中的ORF Finder(https://www.ncbi.nlm.nih.gov/orffinder/)查找蛋白质编码基因,并与长蝽总科中已知的7种线粒体基因组数据进行比对,确定蛋白质编码基因边界位置。
控制区中的串联重复序列通过在线工具Tandem Repeats Finder(http://tandem.bu.edu/trf/trf.html)进行查找,茎环结构使用The mfold Web Server(http://unafold.rna.albany.edu/?q=mfold)(Zuker, 2003)进行预测。
MEGA 6.0(Tamuraetal., 2013)进行统计分析线粒体基因组的核苷酸组成和蛋白质编码基因的密码子使用情况。公式AT skew=(A-T)/(A+T)和GC skew=(G-C)/(G+C)(Perna and Kocher, 1995)计算得到核苷酸组成的偏向性。
1.5 系统发育分析
选取蝽次目内15种线粒体基因组数据重建长蝽总科系统发育关系,其中长蝽总科的7个物种作为内群 (Huaetal., 2008;Lietal., 2013;Lietal., 2016a),蝽总科、红蝽总科和缘蝽总科的8个物种作为外群。地长蝽内的白斑地长蝽Panaorusalbomaculatus导致地长蝽在系统发育树中处于一个奇怪的位置 (Lietal., 2016b),本次研究中并未使用白斑地长蝽线粒体基因组数据(表1)。
依据13个蛋白质编码基因DNA序列,使用贝叶斯分析(Bayesian inference)和最大似然法(Maximum likelihood)重建系统发育树,核苷酸矩阵的替换模型通过jModelTest 2.1.4(Posada, 2008)计算得到。贝叶斯法使用MrBayes 3.12(Huelsenbeck and Ronquist, 2001)运算,共运行1千万代,每隔1000代取一次样,然后舍去前25%。最大似然法使用RAxML 8.2.9(Stamatakis, 2006)运算,Bootstrap replicates值为1000。
2 结果与分析
2.1 大黑毛肩长蝽线粒体基因组结构
大黑毛肩长蝽线粒体基因组是双链闭合环状DNA分子,长17097 bp,GenBank序列号为KY039274,含有13个蛋白质编码基因,22个tRNA基因和2个rRNA基因(图1,表3)。基因排列方式同果蝇一致(Clary and Wolstenholme, 1985)。13个蛋白质编码基因中,4个基因(ND4,ND4L,ND5,ND1)位于N链,其余9个位于J链;22个tRNA基因中,8个基因(tRNA-Gln,tRNA-Cys,tRNA-Tyr,tRNA-Phe,tRNA-His,tRNA-Pro,tRNA-Leu(UAG),tRNA-Val)位于N链,其余14个位于J链;2个rRNA基因全部位于N链。
大黑毛肩长蝽线粒体基因组结构紧凑,共观察到10处基因重叠,重叠总长度是37 bp,其中tRNA-Trp和tRNA-Cys的重叠序列最长,为8 bp。ND4与ND4L间的重叠序列是ATGATAG与Cameron(2014b)报道的重叠序列(ATGATAA)略有不同。同时在该基因组内存在13处基因间隔区,长度范围为1-18 bp,位于ND1和tRNA-Ser(UGA)的间隔区最长,为18 bp。此外,大黑毛肩长蝽线粒体基因组内还存在2个大的非编码区。一个是控制区(图2A),另一个是位于ND6和CytB之间的串联重复区域,TRR4.4(图2B)。

图1 大黑毛肩长蝽线粒体基因组结构Fig.1 Mitochondrial genome map of Neolethaeus assamensis注:箭头表示基因转录的方向。蓝色代表蛋白质编码基因,紫色代表rRNA基因,红色代表tRNA基因,灰色代表大的非编码区。tRNA使用氨基酸缩写命名。黑色代表GC含量。GC斜率绘制为与整个序列的平均GC斜率的偏差。Note: Arrows indicate the orientation of gene transcription. PCGs are shown as blue arrows, rRNA genes as purple arrows, tRNA genes as red arrows and large non-coding regions as gray arrows. tRNAs are named using single-letter amino acid abbreviations. The GC content is plotted using a black sliding window. GC Skew is plotted as the deviation from the average GC skew of the entire sequence.

基因Gene链Strand位置Position反密码子Anticodon大小(bp)Size起始密码子Startcodon终止密码子Stopcodon间隔IntergenicnucleotidestRNA⁃IleJ1-67GAT67tRNA⁃GlnN65-133TTG69-3tRNA⁃MetJ134-202CAT690ND2J203-12041002ATTTAA0tRNA⁃TrpJ1203-1265TCA63-2tRNA⁃CysN1258-1324GCA67-8tRNA⁃TyrN1329-1394GTA664COIJ1397-29351539TTGTAA2tRNA⁃Leu(UAA)J2931-2995TAA65-5COIIJ2997-3678682ATAT-1tRNA⁃LysJ3679-3752CTT740tRNA⁃AspJ3753-3815GTC630
续上表

基因Gene链Strand位置Position反密码子Anticodon大小(bp)Size起始密码子Startcodon终止密码子Stopcodon间隔IntergenicnucleotidesATPase8J3816-3980165ATCTAA0ATPase6J3974-4639666ATGTAA-7COIIIJ4639-5425787ATGT--1tRNA⁃GlyJ5426-5490TCC650ND3J5491-5844354ATTTAA0tRNA⁃AlaJ5847-5913TGC672tRNA⁃ArgJ5914-5978TCG650tRNA⁃AsnJ5981-6053GTT732tRNA⁃Ser(GCU)J6053-6121GCT69-1tRNA⁃GluJ6124-6192TTC692tRNA⁃PheN6198-6266GAA695ND5N6266-79841719ATTTAA-1tRNA⁃HisN7985-8049GTG650ND4N8055-93801326ATGTAA5ND4LN9374-9655282ATTTAG-7tRNA⁃ThrJ9658-9728TGT712tRNA⁃ProN9729-9794TGG660ND6J9797-10285489ATATAA2TRR4410296-1109780210CytBJ11105-122381134ATGTAG7tRNA⁃Ser(UGA)J12237-12306TGA70-2ND1N12325-13251927ATTTAA18tRNA⁃Leu(UAG)N13252-13318TAG67016SrRNAN13319-1459312750tRNA⁃ValN14594-14664TAC71012SrRNAN14665-154397750Controlregion15440-1709716580
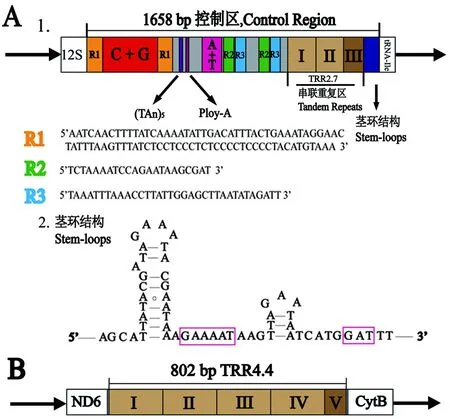
图2 大黑毛肩长蝽线粒体基因组大的非编码区Fig.2 Large non-coding regions in Neolethaeus assamensis mitochondrial genome注:A,控制区;A1,大黑毛肩长蝽线粒体控制区的结构;黄色、绿色和蓝色代表3个不同的重复序列。A2,控制区内预测的茎环结构;红色框代表保守的元件,“G(A)nT”。B,TRR4.4的位置和结构。Note: A, Control region: A1, Structure elements found in the control region of N. assamensis; The yellow, green and blue boxes represent three different repetitive sequences found in the control region. A2, The putative stem-loop structure was found in the control region; The red box regions indicate conserved “G(A)nT” motif. B, Location and structure of TRR4.4.
2.2 控制区结构
控制区是大黑毛肩长蝽线粒体基因组中最大的非编码区,5′端毗邻12S rRNA,3′端邻近tRNA-Ile,长度为1658 bp,AT含量是71.3%。控制区中有如下7类共9个结构显著的区域:(1)长度 429 bp的高GC富含区,GC含量是35.4%;(2)24 bp的(TAn)5结构(TAAAAATAAATAAATAAA AATAAA);(3)10 bp的Poly-A结构(AAAAAAC AAAA);(4)高AT富含区,长56 bp,AT含量为92.9%;(5)长479 bp的串联重复区域(TRR2.7)(Tandem repeat region),包含2个184 bp 的完整重复单元(Ⅰ和Ⅱ)和1个该单元的部分序列(Ⅲ);(6)位于3’端的茎环结构;(7)散布在整个控制区内的3对不同的非串联重复序列(图 3A)。
在3对非串联重复序列中,第一对重复序列(R1)的长度是86 bp,两条序列分别位于高GC富含区的两侧。第二对重复序列(R2)的长度是22 bp,第三对重复序列(R3)的长度是34 bp,R2和R3均位于高AT富含区和串联重复区之间。
2.3 TRR4.4结构
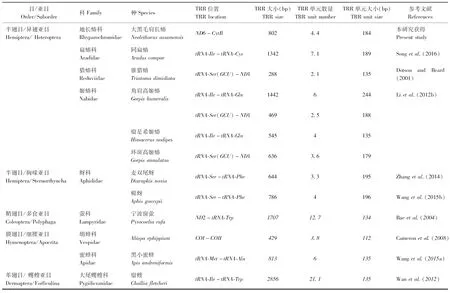
某些昆虫线粒体基因组中,在控制区以外的其他区域能够观察到串联重复序列(表4)。如膜翅目中的黑小蜜蜂Apisandreniformis在tRNA-Met和tRNA-Ala之间存在813 bp的串联重复序列(Wangetal., 2015a),鞘翅目中的宁波窗萤Pyrocoeliarufa在ND2和tRNA-Trp之间存在1707 bp的串联重复序列(Baeetal., 2004)。半翅目扁蝽总科的同扁蝽Araduscompar在tRNA-Ile和tRNA-Cys之间存在1342 bp的串联重复序列(Songetal., 2016)。
在大黑毛肩长蝽线粒体基因组的ND6和CytB之间发现一个非编码的串联重复区域, 即TRR4.4。TRR4.4长802 bp,AT含量是72.2%,包括4个184 bp的重复单元和1个66 bp的部分重复单元。与其他已报道的串联重复序列不同的是,TRR4.4的重复单元与控制区中TRR2.7的重复单元在长度、方向以及核苷酸组成等方面几乎完全一致(图3A)。据我们了解,在昆虫线粒体基因组中,这是首次发现控制区外的串联重复单元与控制区内的串联重复单元一致的现象。

图3 大黑毛肩长蝽线粒体基因组中的TRR4.4和TRR2.7Fig.3 TRR4.4 and TRR2.7 in Neolethaeus assamensis mitochondrial genome注:A,TRR4.4和TRR2.7的重复单元;红色框代表11个碱基的序列位置不同,棕色代表重复单元中相同的序列,绿色代表发生变异的位点。B,TRR4.4和TRR2.7末端预测的二级结构,位于TRR4.4和TRR2.7的核苷酸标为黑色;波浪线代表TRR4.4,TRR2.7内部省略的序列。Note: A, The unit of TRR4.4 and TRR2.7; Red boxes indicate the location of 11 bp sequences in those units, Brown boxes indicate the similar sequences in those units, Green boxes indicate the inconsistent nucleotides. B, Hypothetical secondary structures in the endpoints of TRR4.4 and TRR2.7, in which bold nucleotides locate within TRR4.4 and TRR2.7; The break lines indicate abbreviatory sequences of TRR4.4 and TRR2.7.
串联重复序列是昆虫线粒体基因组控制区内一个较为保守的结构(Zhang and Hewitt, 1997),同时根据我们的了解,在蛋白质编码基因ND6和CytB之间并未有串联重复序列的报道。因此,在大黑毛肩长蝽线粒体基因组中,TRR4.4可能源自控制区内的TRR2.7。串联重复随机丢失模型(TDRL)和重组假说都可以解释这种现象。TDRL模型认为某些基因发生多重拷贝,之后个别基因随机丢失 (Moritzetal., 1987)。依据TDRL模型,首先CytB-TRR2.7的片段产生拷贝,然后突变累积,某些基因失去功能,成为假基因,接着部分拷贝的片段从线粒体基因组中丢失,最终TRR4.4在ND6和CytB之间产生。但是该假设与观察到的现象并不相符,即在线粒体基因组中没有发现CytB-TRR2.7的相关遗迹。重组假说认为重组一般出现在具有自身互补序列(如茎环结构)的重组热点区域(Dowton and Campbell, 2001)。在大黑毛肩长蝽线粒体基因组中,TRR4.4和TRR2.7的末端都重组热点区域邻接(图3B)。从这个意义上说,重组模型可能解释TRR4.4的出现,当然更多的研究还需要进一步开展。
2.4 蛋白质编码基因
大黑毛肩长蝽线粒体基因组的13个蛋白质编码基因全长共11072 bp,除终止子外,共发现3692个密码子。在起始密码子方面,除COI的起始密码子是TTG外,其余12个蛋白编码基因的起始密码子均为ATN;在终止密码子方面,11个蛋白编码基因以TAA或TAG作为终止密码子,而COII和COIII以单个T作为终止密码子,其下游是同链编码的tRNA基因(表5)。
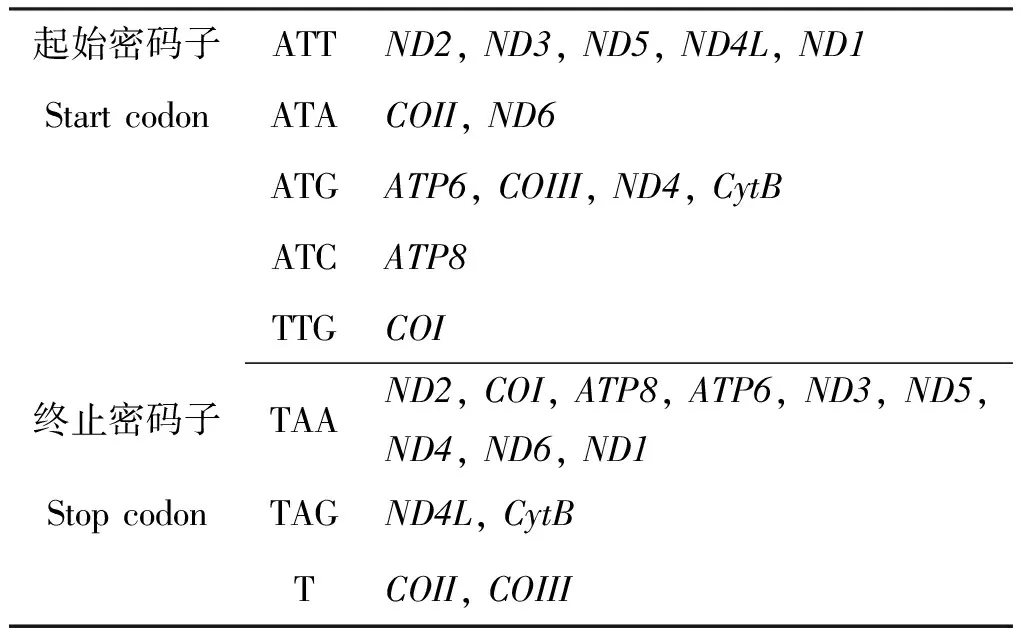
表5 大黑毛肩长蝽线粒体基因组中蛋白质编码基因的起始密码子和终止密码子
2.5 tRNA
大黑毛肩长蝽线粒体基因组共有22个tRNA基因,长度范围从63 bp(tRNA-Trp和tRNA-Asp)到74 bp(tRNA-Lys),tRNA基因的AT含量比较丰富,为76.6%,并且表现出AT偏移。tRNA-Ser(GCU)的二级结构中含有一个DHU臂,并没有出现常见的环状结构,这一现象在悬铃木方翅网蝽Corythuchaciliata(Yangetal., 2013)中同样有报道。因此,22个tRNA全部能够折叠为典型的三叶草二级结构(图4)。
在tRNA的二级结构中观察到绝大多数tRNA的氨基酸接受臂、反密码子臂和反密码子环的长度相对保守,分别为7 bp,5 bp和7个核苷酸,相反DHU环,TψC臂和TψC环的变化范围较大(表6)。此外,共发现24处non-Watson-Crick碱基配对(表7),分别为G-U(19),U-U(2),A-C(2)和C-U(1)。其中,16处发现于N链编码的tRNA,8处发现于J链编码的tRNA。
2.6 rRNA
大黑毛肩长蝽线粒体基因组中,16S rRNA基因长1275 bp,位于tRNA-Leu(UAG)和tRNA-Val之间,其二级结构包含6个结构域(结构域III在节肢动物中缺失)和44个茎环结构(图5)。结构域I、II和VI的变异幅度较大(Huaetal., 2008;Lietal., 2013)。H991的序列变异大(Lietal., 2012a;Songetal., 2013;Wangetal., 2014),缺乏相应参考,其二级结构通过RNAstructure 5.8(Reuter and Mathews, 2010)和Mfold(Zuker, 2003)预测得到。参照Buckley等(2000)的模型,H2347折叠成3 bp的茎环结构。
12S rRNA基因长775 bp,位于tRNA-Val和控制区之间,其二级结构包含3个结构域和28个茎环结构(图6),其中结构域III较为保守。H769的二级结构折叠形成2个茎环结构,即H769a和H769b。
2.7 核苷酸组成和密码子使用
大黑毛肩长蝽线粒体基因组的AT含量是73.5%,所有基因都富含AT(表8),其中rRNA基因的AT含量最高,为76.9%。在13个蛋白质编码基因中,ND2的AT含量最高,为78.5%;COI的AT含量最低,为67.6%。线粒体基因组中核苷酸组成在不同链间是不对称的 (Perna and Kocher, 1995)。J链编码的蛋白质基因和tRNA基因均为AT偏移和CG偏移。N链编码的蛋白质基因、tRNA基因和rRNA基因均为TA偏移和GC偏移。

图4 大黑毛肩长蝽线粒体基因组中tRNA的二级结构Fig.4 Putative secondary structures of tRNAs in Neolethaeus assamensis mitochondrial genome注:tRNAs 使用相应的氨基酸缩写表示。典型的碱基配对使用短线连接,GU配对使用星号表示,其他非典型配对使用空心圆表示。Note: The tRNAs are labeled with the abbreviations of their corresponding amino acids. Inferred Watson-Crick bonds are illustrated by lines, GU bonds by asterisk, and the other non-Watson-Crick interactions are illustrated by hollow circles.

tRNA链Strand氨基酸接受臂(bp)AAstem反密码子臂(bp)ACstem反密码子环(nucleotide)ACloopDHU臂(bp)DHUstemDHU环(nucleotide)DHUloopTψC臂(bp)TψCstemTψC环(nucleotide)TøCloop可变环(nucleotide)VariableLooptRNA⁃AlaJ75744564tRNA⁃ArgJ75743544tRNA⁃AsnJ757394105tRNA⁃AspJ75746344tRNA⁃GluJ75747474tRNA⁃GlyJ75737374tRNA⁃IleJ75736395tRNA⁃Leu(UAA)J75736544tRNA⁃LysJ757311575tRNA⁃MetJ74745575tRNA⁃Ser(GCU)J75734674tRNA⁃Ser(UGA)J75745574tRNA⁃ThrJ757473114tRNA⁃TrpJ75745434tRNA⁃CysN75747534tRNA⁃GlnN73945575tRNA⁃HisN75746444tRNA⁃Leu(UAG)N75738544tRNA⁃PheN75747554tRNA⁃ProN75745464tRNA⁃TyrN757310274tRNA⁃ValN75745574

表7 大黑毛肩长蝽线粒体基因组tRNA中非典型的碱基对

图5 大黑毛肩长蝽线粒体基因组中16S rRNA的二级结构Fig.5 Predicted secondary structure of the 16S rRNA in Neolethaeus assamensis mitochondrial genome注:灰色代表长蝽总科中高度保守的区域。罗马数字代表结构域。典型的碱基配对使用短线连接,GU配对使用星号表示,其他非典型配对使用空心圆表示。Note: Grey regions indicate high conserved sites in Lygaeoidea. Roman numerals denote the conserved domain structure. Inferred Watson-Crick bonds are illustrated by lines, GU bonds by asterisk and the other non-Watson-Crick interactions are represented by hollow circles.

图6 大黑毛肩长蝽线粒体基因组中12S rRNA的二级结构Fig.6 Predicted secondary structure of the 12S rRNA in Neolethaeus assamensis mitochondrial genome注:灰色代表长蝽总科中高度保守的区域。罗马数字代表结构域。典型的碱基配对使用短线连接,GU配对使用星号表示,其他非典型配对使用空心圆表示。Note: Grey regions indicate relatively conserved sites in Lygaeoidea. Roman numerals denote the conserved domain structure. Inferred Watson-Crick bonds are illustrated by lines, GU bonds by asterisk and the other non-Watson-Crick interactions are represented by hollow circle.
蛋白编码基因密码子的使用表现出极大的AT不均质性,密码子第三位的AT含量最高,是83.4%,而密码子第一、二位的AT含量仅为68.4%和66.7%。大黑毛肩长蝽线粒体基因组中使用频率最频繁的4种密码子全部由A、T组成:TTA(299),TTT(271),ATA(270)和ATT(261)。J链编码的蛋白质基因中,NNA在四倍简并密码子中占优势,NNA和NNU在双重简并密码子中占优势;N链编码的蛋白质基因中,NNU在四倍简并密码子中占优势,NNA和NNU在大多数的双重简并密码子中占优势(图7)。
2.8 系统发育关系
贝叶斯分析和最大似然法分析产生的系统发育树拓扑结构一致(图8)。系统发育与Xie等(2005)和Hua等(2008)一致,支持(缘蝽总科+长蝽总科)的单系性。地长蝽位于长蝽总科的基部分支,与长蝽总科其他类群形成姐妹群关系,这一结果与Henry(1997)中依据形态学证据得到的结论相符;但是该结论与Li等(2016b)基于分子数据得到的结果不同,后者的结果有可能为假阴性。因此,需要获取长蝽总科中更多类群的基因组信息,以便于长蝽总科系统发育关系的进一步探讨。
3 结论与讨论
大黑毛肩长蝽线粒体基因组大小为17097 bp,在已知的6个长蝽总科昆虫线粒体基因组(全序列)中长度最长,长度最小的是长蝽科Lygaeidae的桦穗长蝽Kleidocerysresedae,为14688 bp(Lietal., 2016a)。控制区是长蝽总科线粒体基因组大小变异的主要来源,一般控制区的大小与线粒体基因组大小呈现正相关关系,如大黑毛肩长蝽线粒体基因组的控制区大小为1658 bp,而桦穗长蝽线粒体基因组的控制区长度仅为220 bp。

表8 大黑毛肩长蝽线粒体基因组核苷酸组成分析

图7 大黑毛肩长蝽线粒体基因组中每个氨基酸的同义密码子使用率Fig.7 Percentage of synonymous codon usage of each amino acid in the Neolethaeus assamensis mitochondrial genome注:X轴上为密码子家族。Note: Codon families are provided on the x-axis.

图8 基于线粒体基因组蛋白质编码基因所得到的地长蝽科系统发育地位Fig.8 Phylogenetic position of Rhyparochromidae in Lygaeoidea inferred from the sequences of PCGs in mitochondrial genome注:贝叶斯分析和最大似然法分析具有一致的拓扑结构。节点处上方的数字代表贝叶斯后验概率,下方代表bootstrap值。Note: Bayesian analyses and Maximum Likelihood show the same topology. Numbers at the nodes are Bayesian posterior probabilities (up) and Maximum Likelihood bootstrap values (down).
大黑毛肩长蝽线粒体基因组的AT含量为73.5%,在6个长蝽总科昆虫线粒体基因组(全序列)中AT含量最低,AT含量最高的是束长蝽科Malcidae的瓜束长蝽Malcusinconspicuus,为77.8%(Huaetal., 2008)。昆虫线粒体基因组控制区由于富含AT碱基,被称为AT丰富区(Stormo, 1982)。研究中发现在长蝽总科某些类群中控制区并不是AT含量最丰富的区域,例如桦穗长蝽、豆突眼长蝽、大黑毛肩长蝽的控制区都不是AT含量最高的区域。
在长蝽总科昆虫线粒体基因组中绝大部分蛋白质编码基因使用ATN和TTG作起始密码子。起始密码子中,使用最频繁的是ATG,而ATC出现频次远远低于其他几种起始密码子。所有线粒体基因COI的起始密码子是TTG。终止密码子方面,大部蛋白质基因使用的终止密码子是TAA或TAG,终止密码子为TA或单个T的情况较少。而且以单个T作为终止密码子的现象多发生在COII和COIII中。
大黑毛肩长蝽线粒体基因组编码37个基因,基因排列顺序与果蝇一致。在线粒体基因组中,tRNA-Ser(GCU)的二级结构中含有一个DHU臂,22个tRNA全部能够折叠为典型的三叶草二级结构。大黑毛肩长蝽线粒体基因组最显著的特征是ND6和CytB之间的串联重复区域,TRR4.4的重复单元与控制区中TRR2.7的重复单元在长度、方向以及核苷酸组成等方面几乎完全一致。系统发育结果支持地长蝽科位于长蝽总科的基部位置,与长蝽总科其他类群形成姐妹群这一观点,但是该结果与Li等(2016b)基于白斑地长蝽的分子数据得到的结论不一致,为确定地长蝽科的系统发育地位,未来需要获取长蝽总科中更多类群的基因组信息。
References)
Bae JS, Kim I, Sohn HD,etal. The mitochondrial genome of the firefly,Pyrocoeliarufa: Complete DNA sequence, genome organization, and phylogenetic analysis with other insects [J].MolecularPhylogeneticsandEvolution, 2004, 32 (3): 978-985.
Bernt M, Donath A, Jühling F,etal. MITOS: Improved de novo metazoan mitochondrial genome annotation [J].MolecularPhylogeneticsandEvolution, 2013, 69 (2): 313-319.
Boore JL. Animal mitochondrial genomes [J].NucleicAcidsResearch, 1999, 27 (8): 1767-1780.
Boore JL. Complete mitochondrial genome sequence of the polychaete annelidPlatynereisdumerilii[J].MolecularBiologyandEvolution, 2001, 18 (7): 1413-1416.
Buckley TR, Simon C, Flook PK,etal. Secondary structure and conserved motifs of the frequently sequenced domains IV and V of the insect mitochondrial large subunit rRNA gene [J].InsectMolecularBiology, 2000, 9 (6): 565-580.
Cameron SL. Insect mitochondrial genomics: Implications for evolution and phylogeny [J].AnnualReviewofEntomology, 2014a, 59: 95-117.
Cameron SL. How to sequence and annotate insect mitochondrial genomes for systematic and comparative genomics research [J].SystematicEntomology, 2014b, 39 (3): 400-411.
Cameron SL, Whiting MF. The complete mitochondrial genome of the tobacco hornworm,Manducasexta, (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths [J].Gene, 2008, 408 (1): 112-123.
Cameron SL, Dowton M, Castro LR,etal. Mitochondrial genome organization and phylogeny of two vespid wasps [J].Genome, 2008, 51 (10): 800-808.
Cannone JJ, Subramanian S, Schnare MN,etal. The comparative RNA web (CRW) site: An online database of comparative sequence and structure information for ribosomal, intron, and other RNAs [J].BMCBioinformatics, 2002, 3 (1): 2.
Clary DO, Wolstenholme DR. The mitochondrial DNA molecule ofDrosophilayakuba: Nucleotide sequence, gene organization, and genetic code [J].JournalofMolecularEvolution, 1985, 22 (3): 252-271.
Dotson EM, Beard CB. Sequence and organization of the mitochondrial genome of the Chagas disease vector,Triatomadimidiata[J].InsectMolecularBiology, 2001, 10 (3): 205-215.
Dowton M, Campbell NJ. Intramitochondrial recombination-is it why some mitochondrial genes sleep around [J].TrendsinEcology&Evolution, 2001, 16 (6): 269-271.
Gillespie JJ, Johnston JS, Cannone JJ,etal. Characteristics of the nuclear (18S, 5.8 S, 28S and 5S) and mitochondrial (12S and 16S) rRNA genes ofApismellifera(Insecta: Hymenoptera): Structure, organization, and retrotransposable elements [J].InsectMolecularBiology, 2006, 15 (5): 657-686.
Hall TA. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT [J].NucleicAcidsSymposiumSeries, 1999, 41 (41): 95-98.
Henry TJ. Phylogenetic analysis of family groups within the infraorder Pentatomomorpha (Hemiptera: Heteroptera), with emphasis on the Lygaeoidea [J].AnnalsoftheEntomologicalSocietyofAmerica, 1997, 90 (3): 275-301.
Henry T. Heteroptera, Chapter 10. In: Foottit RG, Adler PH, eds. Insect Biodiversity: Science and Society [M]. Oxford: Blackwell Publishing Ltd., 2009: 223-263.
Hua J, Li M, Dong P,etal. Comparative and phylogenomic studies on the mitochondrial genomes of Pentatomomorpha (Insecta: Hemiptera: Heteroptera) [J].BMCGenomics, 2008, 9 (1): 1.
Huelsenbeck JP, Ronquist F. MrBayes: Bayesian inference of phylogenetic trees [J].Bioinformatics, 2001, 17: 754-755.
Li H, Liu H, Shi A,etal. The complete mitochondrial genome and novel gene arrangement of the unique-headed bugStenopiratessp. (Hemiptera: Enicocephalidae) [J].PLoSONE, 2012a, 7 (1): e29419.
Li H, Liu H, Song F,etal. Comparative mitogenomic analysis of damsel bugs representing three tribes in the family Nabidae (Insecta: Hemiptera) [J].PLoSONE, 2012b, 7 (9): e45925.
Li T, Gao C, Cui Y,etal. The complete mitochondrial genome of the stalk-eyed bugChauliopsfallaxScott, and the monophyly of Malcidae (Hemiptera: Heteroptera) [J].PLoSONE, 2013, 8 (2): e55381.
Li T, Yi W, Zhang H,etal. Complete mitochondrial genome of the birch catkin bugKleidocerysresedaeresedae, as the first representative from the family Lygaeidae (Hemiptera: Heteroptera: Lygaeoidea) [J].MitochondrialDNAPartA, 2016a, 27 (1): 618-619.
Li T, Yang J, Li Y,etal. A mitochondrial genome of Rhyparochromidae (Hemiptera: Heteroptera) and a comparative analysis of related mitochondrial genomes [J].ScientificReports, 2016b, 6: 35175.
Lowe TM, Eddy SR. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence [J].NucleicAcidsResearch, 1997, 25 (5): 955-964.
Moritz C, Dowling TE, Brown WM. Evolution of animal mitochondrial DNA: Relevance for population biology and systematic [J].AnnualReviewofEcologyandSystematics, 1987, 18 (1): 269-292.
Perna NT, Kocher TD. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes [J].JournalofMolecularEvolution, 1995, 41 (3): 353-358.
Posada D. jModelTest: Phylogenetic model averaging [J].MolecularBiologyandEvolution, 2008, 25 (7): 1253-1256.
Reineke A, Karlovsky P, Zebitz CPW. Preparation and purification of DNA from insects for AFLP analysis [J].InsectMolecularBiology, 1998, 7 (1): 95-99.
Reuter JS, Mathews DH. RNAstructure: Software for RNA secondary structure prediction and analysis [J].BMCBioinformatics, 2010, 11 (1): 1.
Saccone C, De Giorgi C, Gissi C,etal. Evolutionary genomics in Metazoa: The mitochondrial DNA as a model system [J].Gene, 1999, 238 (1): 195-209.
Schuh RT, Slater JA. True Bugs of the World (Hemiptera: Heteroptera): Classification and Natural History [M]. New York: Cornell University Press, 1995: 258-264.
Shi AM, Li H, Bai XS,etal. The complete mitochondrial genome of the flat bugAradacanthiaheissi(Hemiptera: Aradidae) [J].Zootaxa, 2012, 3238 (1): 23-38.
Song F, Li H, Shao R,etal. Rearrangement of mitochondrial tRNA genes in flat bugs (Hemiptera: Aradidae) [J].ScientificReports, 2016, 6: 25725.
Song W, Li H, Song F,etal. The complete mitochondrial genome of a tessaratomid bug,Eusthenescupreus(Hemiptera: Heteroptera: Pentatomomorpha: Tessaratomidae)[J].Zootaxa, 2013, 3620 (2): 260-272.
Stamatakis A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models [J].Bioinformatics, 2006, 22 (21): 2688-2690.
Stormo GD, Schneider TD, Gold LM. Characterization of translational initiation sites inE.coli[J].NucleicAcidsResearch, 1982, 10 (9): 2971-2996.
Tamura K, Stecher G, Peterson D,etal. MEGA6: Molecular evolutionary genetics analysis version 6.0 [J].MolecularBiologyandEvolution, 2013, 30 (12): 2725-2729.
Wolstenholme DR. Genetic novelties in mitochondrial genomes of multicellular animals [J].CurrentOpinioninGenetics&Development, 1992, 2 (6): 918-925.
Wan X, Kim MI, Kim MJ,etal. Complete mitochondrial genome of the free-living earwig,Challiafletcheri(Dermaptera: Pygidicranidae) and phylogeny of Polyneoptera [J].PLoSONE, 2012, 7 (8): e42056.
Wang P, Li H, Wang, Y,etal. The mitochondrial genome of the plant bugApolyguslucorum(Hemiptera: Miridae): Presently known as the smallest in Heteroptera [J].InsectScience, 2014, 21 (2): 159-173.
Wang AR, Kim MJ, Lee JY,etal. The mitochondrial genome of the black dwarf honey bee,Apisandreniformis(Hymenoptera: Apidae) [J].MitochondrialDNA, 2015a, 26 (6): 914-916.
Wang Y, Chen J, Jiang LY,etal. Hemipteran mitochondrial genomes: Features, structures and implications for phylogeny [J].InternationalJournalofMolecularSciences, 2015b, 16 (6): 12382-12404.
Xie Q, Bu W, Zheng L. The Bayesian phylogenetic analysis of the 18S rRNA sequences from the main lineages of Trichophora (Insecta: Heteroptera: Pentatomomorpha) [J].MolecularPhylogeneticsandEvolution, 2005, 34 (2): 448-451.
Yang W, Yu W, Du Y. The complete mitochondrial genome of the sycamore lace bugCorythuchaciliata(Hemiptera: Tingidae) [J].Gene, 2013, 532 (1): 27-40.
Yuan ML, Zhang QL, Guo ZL. The complete mitochondrial genome ofCorizustetraspilus(Hemiptera: Rhopalidae) and phylogenetic analysis of Pentatomomorpha[J].PLoSONE, 2015, 10 (6): e0129003.
Zhang B, Ma C, Edwards O,etal. The mitochondrial genome of the Russian wheat aphidDiuraphisnoxia: Large repetitive sequences betweentrnEandtrnFin aphids [J].Gene, 2014, 533 (1): 253-260.
Zhang DX, Hewitt GM. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies [J].BiochemicalSystematicsandEcology, 1997, 25 (2): 99-120.
Zheng LY, Zou HG. Chinese Bug Identification Manual [M]. Beijing: Science Press, 1981: 116-215. [郑乐怡, 邹环光. 中国蝽类鉴定手册(半翅目:异翅亚目长蝽科)[M]. 北京: 科学出版社, 1981: 116-215] Zuker M. Mfold web server for nucleic acid folding and hybridization prediction [J].NucleicAcidsResearch,2003, 31 (13): 3406-3415.
(Hemiptera: Heteroptera: Rhyparochromidae) and the implication for the phylogenetic position of Rhyparochromidae in Lygaeoidea
GUO Peng-Lei1, WU Yan-Zhuo1, WANG Yan-Hui1, WU Hao-Yang1, LI Teng2, LI Jun-Lan3, BU Wen-Jun1, XIE Qiang4*
(1. College of Life Sciences, Nankai University, Tianjin 300071, China; 2. College of Life Sciences, Lanzhou University, Lanzhou 730000, China; 3. College of Life Sciences, Inner Mongolia University, Hohhot 010021, China; 4. Key Laboratory of Biodiversity Dynamics and Conservation of Guangdong Higher Education Institute, The Museum of Biology, School of Life Sciences, Sun Yat-sen University, Guangzhou 510275, China)
To better understand the genome-level characteristics of Rhyparochromidae, the complete mitochondrial genome ofNeolethaeusassamensisDistant, 1901 (Hemiptera: Heteroptera: Rhyparochromidae: Lethaeini) was sequenced and analyzed. It is a typical double-strand circular molecule with 17097 bp in length, including 13 protein coding genes (PCGs), 22 transfer RNA (tRNA) genes and 2 ribosomal RNA (rRNA) genes. The gene arrangement is similar to that ofDrosophilayakuba. The complete mitochondrial genome has two large non-coding regions, including the control region and a tandem repeat region, TRR4.4, betweenND6 andCytB. The control region has nine structural elements categorized as seven different types: a stem-loops region, three non-tandem repeat sequences, and five other motifs. TRR4.4 is 802 bp in length, which consisted of four identical DNA sequences (184 bp) and a partial copy of such sequence (66 bp). The unit of the TRR4.4 is similar to that of the TRR2.7 in the control region in size, direction, and nucleotides composition. All tRNAs can be folded into the classic clover-leaf secondary structure. The secondary structures of rRNAs have also been predicted. The 16S rRNA is comprised of six domains (domain III is absent in arthropods) and 44 helices, while the 12S rRNA consists of three domains and 28 helices. Phylogenetic analyses based on the mitochondrial genomes of 15 species of Pentatomomorpha showed that Rhyparochromidae is the basal lineage in Lygaeoidea.
Mitochondrial genome;Neolethaeusassamensis; tandem repeat; recombination; phylogenetic analyses
国家自然科学基金(31572242)
郭鹏磊,男,1991年生,硕士研究生,研究方向为昆虫分子系统学,E-mail: NKGPL2014@126.com
*通讯作者Author for correspondence,E-mail:xieq8@mail.sysu.edu.cn
Received: 2017-02-20;接受日期Accepted: 2017-03-07
Q963;Q961;S433
A
1674-0858(2017)02-0314-18
郭鹏磊,吴燕卓,王艳会,等.大黑毛肩长蝽(半翅目:异翅亚目:地长蝽科)线粒体基因组及地长蝽科系统发育地位探讨[J].环境昆虫学报,2017,39(2):314-331.
