密度泛函理论研究银上吸附对巯基吡啶的SERS化学增强效应
2017-05-10吴元菲李明雪周剑章吴德印田中群
吴元菲 李明雪 周剑章 吴德印 田中群
(厦门大学化学化工学院化学系,固体表面国家重点实验室,能源材料化学协同创新中心,福建厦门361005)
密度泛函理论研究银上吸附对巯基吡啶的SERS化学增强效应
吴元菲 李明雪 周剑章 吴德印*田中群*
(厦门大学化学化工学院化学系,固体表面国家重点实验室,能源材料化学协同创新中心,福建厦门361005)
基于密度泛函理论计算和拉曼光谱理论分析,我们研究了对巯基吡啶(4MPY)分子的拉曼光谱和其在银上的表面增强拉曼光谱(SERS),并进一步探讨了SERS与界面吸附结构、异构化、质子化和氢键作用以及低能激发态的关系。首先,我们对两种分子异构体的相对稳定性和拉曼光谱进行了理论分析。在此基础上,进而研究了该分子与不同银簇作用时的拉曼光谱,结果表明,4MPY以巯基硫与银簇作用形成强的Ag―S键,导致拉曼光谱的线型不依赖于所选银簇的大小。接着我们考虑了吡啶氮端作用的两种情况。(1)当4MPY-银簇复合物同时以吡啶氮与水簇或水合质子簇形成氢键时,结果表明吡啶环的部分振动频率随氢键和质子化发生蓝移。(2)当考虑吡啶氮与银簇作用时,吡啶环三角畸变振动发生蓝移。上述情况不仅解释了实验观测的振动频率变化,而且表明了化学环境改变对相对拉曼强度的影响。最后,我们计算了当对巯基吡啶分子以单端或双端与银簇作用,在考虑激发光与低能激发态的能量匹配时,拉曼光谱强度与低能激发态的关系。计算结果表明,在双端吸附构型下,与吡啶氮成键的银簇受激发产生电荷转移态,不仅导致吡啶环v12、v1和v8a振动的拉曼信号增强,而且选择性地增强吡啶环C―H面内对称弯曲振动v9a的拉曼信号。
表面增强拉曼光谱;密度泛函理论;电荷转移机理;对巯基吡啶;银
1 引言
表面增强拉曼光谱(SERS)在币族金属纳米结构表面具有高的检测灵敏度,可用于研究低浓度的表面物种鉴定和表征。SERS光谱技术不仅在空间分辨率上有提升,而且在能量和时间分辨上也有所提高,这为研究界面吸附的探针分子提供了重要的指纹信息1,2。人们目前普遍认为SERS增强效应主要来自电磁增强机理和化学增强机理。电磁增强机理主要由币族金属纳米结构的表面等离激元共振(SPR)效应决定,与纳米结构表面的粗糙度和分布有关。如果SERS基底由纳米粒子组成,则增强效应与纳米粒子的材料光学性质、大小、形状以及聚集状态有关3。化学增强机理与吸附分子的几何结构、电子结构、分子与基底的成键作用和界面环境密切相关。它主要是由于直接的化学吸附作用,或与吸附物与基底之间形成新的电荷转移态相关4。依据表面选律,一般情况下,电磁增强机理对相同对称性振动模的相对SERS强度影响较小,而化学增强直接影响吸附分子和拉曼谱带的相对强度,同时与SERS强度相关的化学增强因素也会直接影响探针分子的振动频率5。与探针分子在气相或溶液中的拉曼光谱相比,当探针分子吸附于界面,其SERS光谱可能发生显著变化,这常常阻碍了认识SERS光谱的本质6,7。
对巯基吡啶(4MPY)是SERS研究中重要的模型分子,通常具有一定的化学稳定性8,9。但在溶液或固体中可能存在两种异构体,即苯式结构和醌式结构的平衡2,3。当4MPY的巯基在银电极表面形成强的化学吸附态时,其化学结构可很好地被用于化学增强机理研究10,11。同时,利用其构筑特殊的界面自组装分子层所具有强的拉曼信号,可方便用于表面分析6,7,12。前人研究该分子在银电极上随电位调制测得不同激发波长下的SERS光谱时,发现随激发波长增加,光谱峰值随电位负移,由此提议电荷转移方向是从金属到分子13。然而,4MPY在表面的吸附结构并不确定14,15。由于吡啶环上N原子在低的pH值下可发生质子化,从而可用SERS研究pH与拉曼光谱的关系,探测金属-溶液界面吸附分子的p Ka值,如之前的研究将该分子作为界面pH敏感的探针分子16和氢键手性传递探针17,而目前很少讨论氮原子与金属的作用11,12。在银纳米结构表面,吡啶分子的N原子与银表面有一定的化学吸附,这给我们启发,4MPY是否可能以巯基硫和吡啶环氮两端同时吸附在表面,形成双端吸附构型。
在理论研究方面,密度泛函理论(DFT)计算已用于4MPY分子吸附在金和银纳米结构表面的SERS光谱的分析17-26。基于非谐性考虑,Respondek和Benoit18对4MPY的振动谱峰进行了详细的分析。Wang等17从分子间氢键作用考虑,对拉曼谱峰进行了分析。Mo研究组等25,26较早对4MPY分子本身及其与银簇作用的复合物进行了结构优化和振动谱峰指认。Sun等19基于DFT计算研究了4MPY以巯基硫与TiO2簇作用,探讨电荷转移对4MPY的拉曼光谱影响。最近,Birke和Lombardi20基于Ag13簇模型从静态和动态的极化率分析了4MPY的SERS光谱,Liu等21以Ag20簇模拟纳米结构表面吸附,他们分别进行了巯基硫和吡啶氮与银作用的SERS光谱计算。Iida等22基于平面波方法以4MPY分子作为探针分子,进行了电化学电位对SERS光谱的理论计算。目前所有这些研究均主要考虑4MPY分子以巯基硫吸附在银或金表面,未考虑溶剂化效应或吡啶环氮原子与溶剂分子的氢键作用,也忽略了巯基和吡啶环氮同时吸附所形成的双端吸附构型。
在本工作中,我们采用金属簇模型方法,详细地探讨4MPY探针分子的异构化,以多种方式与银簇作用以及界面溶剂对探针分子的拉曼光谱影响,分析了SERS光谱与界面吸附结构的关系。我们也对4MPY和它的异构体拉曼光谱进行分析和归属,并与固体粉末的拉曼光谱进行比较,检查DFT方法用在计算该分子结构和拉曼光谱时的合理性。基于对分子结构和拉曼光谱的研究,进一步比较当该探针分子通过巯基硫与不同银簇作用后,强的化学吸附作用对其拉曼光谱的影响。最后,在单端和双端结构下,我们更进一步分析低能激发态和电荷转移机理对4MPY的拉曼光谱强度的影响,进而确定SERS与表面吸附结构的关系。
2 计算方法
为了建立4MPY分子在银上的吸附结构,我们考虑了4MPY的两种异构体PYSH和PYSNH,并采用金属簇模型方法模拟金属表面活性位,进而用分子与金属簇的作用描述分子在银表面活性位上吸附作用。图1示出分子及其与银簇形成复合物的结构。簇模型方法不仅适合考虑强的化学吸附体系,而且便于计算吸附分子激发态,并模拟其表面振动光谱5。4MPY可在银表面发生强的化学吸附,通过巯基与银形成强的Ag―S键23。在银上所形成的Ag―S键可能在不同的表面位,如顶位、桥位和穴位等。我们以前的研究表明,芳香环的巯基硫偏好吸附在桥位或穴位23。除了考虑4MPY以巯基硫的单端吸附构型外,因用于SERS研究的金属纳米结构具有一定的粗糙度,且粗糙度常大于分子尺度,因此,我们建议在银上4MPY有可能以吡啶环上氮原子孤对与银表面作用,如图1B所示,我们也考虑了4MPY分子以吡啶氮与银簇作用的情况。
对上述不同的吸附构型,我们采用杂化密度泛函B3LYP方法进行平衡结构计算。对C、N或H原子,计算所采用全电子Gauss基组6-311+G**,而对Ag原子,采用LANL2DZ基组和相应的相对论赝势。我们之前的研究表明该计算水平不仅能很好预测优化结构,而且也可以得到合理的振动频率和拉曼光谱强度5,23。

图1 (A)4MPY分子的质子化、去质子化和异构化平衡以及(B)4-M PY吸附在银纳米结构上的4-M PY-银簇结构模型Fig.1(A)Equilibrium of protonation,deprotonation, isomerization of 4MPY aswellas(B)structuremodelings of4MPY-silver clustersof 4MPY adsorbed on silver nanostructures
4MPY分子可能以质子化形式或两性离子形式吸附于银纳米结构表面,界面溶剂会显著影响其SERS光谱。为此,我们考虑了溶剂水对表面吸附4MPY分子的SERS光谱影响。这里我们采用显性(H2O)8水簇与吡啶氮成氢键,并采用极化连续模型(SMD)进行计算。对于吡啶环上氮原子的质子化,我们采用水合质子H9O4+模型,并在此基础上使用SMD模型进行结构优化和拉曼光谱计算。在不共振的情况下,计算的拉曼强度采用激发波长为632.8 nm。
为考虑电荷转移增强机理,我们采用含时密度泛函理论(TD-DFT)方法计算了4MPY-银簇复合物的低能激发态,并通过分析相关跃迁轨道、跃迁能和振子强度,鉴别出SERS增强效应相关的电荷转移态。然后基于预共振拉曼光谱理论5,当拉曼激发光能量与低能电荷转移态的激发能接近时,考虑了共振效应计算的吸附4MPY的SERS光谱的影响。
3 结果与讨论
3.1 4MPY异构体的相对稳定性和拉曼光谱
表1列出B3LYP计算在气相和溶剂化条件下两种异构体的相对能量和自由能之差。在气相中,中性分子PYSH比两性离子PYSNH略微稳定,能差仅为6.20 kJ·mol-1。如考虑零点能校正和热容的影响,在101.325 kPa和298.15 K的情况下,两种异构体的相对Gibbs自由能差为19.01 kJ·mol-1。依据Boltzmann分布,这表明在常温气相中4MPY应主要以中性分子形式存在。当考虑水的溶剂化作用后,因PYSNH的偶极矩明显大于PYSH的偶极矩,PYSNH变得稳定。以水为溶剂,采用SMD模型计算,PYSNH比PYSH能量低31.19 kJ·mol-1。在室温下,PYSNH的Gibbs自由能比PYSH低16.87 kJ·mol-1。这表明在水中,4MPY主要以两性离子PYSNH存在。这表明两种异构体的相对稳定性极其依赖于分子所处的状态和溶剂化条件。
4MPY分子有12个原子和30个基频振动模。分子骨架有C2v对称性,则其振动模可分为四类不可约表示,且所有振动模均具有拉曼活性。目前4MPY的气相结构和拉曼光谱仍未见报道。虽然在固体粉末8和中性水溶液中拉曼光谱已有报道24,25,但由于可能存在异构化问题,之前文献对谱峰仅限于依据PYSH异构体的振动谱分析进行归属18,26。
图2是在气相和采用SMD溶剂化模型时计算两种异构体PYSH和PYSNH的拉曼光谱。图2A计算PYSH异构体的拉曼光谱,它的强峰主要为989、1103和1578 cm-1。这些峰分别是吡啶环的三角畸变v12、环呼吸振动v1以及平行于主轴的C―C对称伸缩振动v8a。其次,频率由低到高,依次来自C―S伸缩和吡啶环v6a耦合的399和698 cm-1,吡啶环畸变v6b的663 cm-1,CSH弯曲的921 cm-1、吡啶环的C―H面内弯曲v9a振动的1219和反对称C―C和C―N伸缩的1246 cm-1。
图2B是在气相和考虑溶剂化后计算PYSNH的拉曼光谱。由于异构化,PYSNH与PYSH在拉曼光谱上显著不同。首先,与CSH弯曲振动相关的拉曼谱峰921 cm-1应消失,而与CNH面内弯曲振动频率出现在1213 cm-1。其次,由于这种异构化,N―H键面外弯曲振动基频峰应出现在700 cm-1,但其拉曼强度很弱。最后,PYSNH的最强拉曼谱峰为978 cm-1,主要来自吡啶环三角畸变v12模。较强的拉曼谱峰还有C―S伸缩和环畸变v6a的耦合振动峰421和719 cm-1,吡啶环畸变v6b峰648 cm-1,C―N对称伸缩有关的1034 cm-1以及C―C伸缩和C―H面内弯曲的1373、1449和1479 cm-1。与PYSH强的拉曼谱峰相比,PYSNH的v8a谱峰频率增大到1622 cm-1,这主要是PYSNH的N位质子化后,吡啶环由芳香性向醌式结构转化,平行于主轴的C―C键趋向于定域化强的双键。在相同的条件下,PYSNH的拉曼强度比PYSH的略强。

表1 UB3LYP/6-311+G**水平计算相对能量和相对Gibbs自由能Table1 Relativeenergiesand relativeGibbs freeenergies calculated at the UB3LYP/6-311+G**level

图2在B3LYP/6-311+G**水平计算气相(弱线)和溶剂化模型(强线)的模拟拉曼光谱Fig.2 Simu lated Raman spectra in gasphase(weak curve)and the solvationm odel(strong curve)calculated at the B3LYP/6-311+G**level(A)PYSH,(B)PYSNH.TheRaman bandsareexpanded by using the Lorentzian line shapew ith a linew idth of 10 cm-1at the excitationwavelength 632.8 nm.coloronline
图3 是4MPY分子间以氢键作用的二聚体、四聚体、五聚体以及六聚体的优化结构和相应的计算拉曼光谱。晶体结构测定表明4MPY分子主要以两性离子PYSNH存在,且PYSNH之间以氢键结构成链状或环状结构27-29。图3A是4MPY低聚体的优化结构,结果表明4MPY分子间通过氢键作用,且为PYSNH异构体。在结构上,每个PYSNH的N―H键与另一个PYSNH的S原子成氢键作用,N―H键长约为0.1028 nm,H…S距离约为0.2173-0.2194 nm。在气相中,这种氢键作用较强,计算平均氢键能为50.24 kJ·mol-1。考虑水的溶剂化作用,氢键H…S距离增加到0.2240-0.2332 nm,而计算的平均氢键能减小到13.82 kJ· mol-1。图3B是四种多聚体结构的计算拉曼光谱。在计算拉曼光谱方面,不论在频率上还是在相对拉曼强度上,这些低聚体的计算拉曼光谱之间相差较小,且与实验测定4MPY固体粉末的拉曼光谱相吻合8。

图3 B3LYP/6-311+G**水平计算PYSNH异构体的二聚体、四聚体、五聚体和六聚体的优化结构(A)和模拟拉曼光谱(B)Fig.3 Optim ized structu res(A)and sim ulated Ram an spectra(B)of d im er,tetram er,pentam er,and hexamer of PYSNH calcu lated at the B3LYP/6-311+G**levelThe Raman bandsare expanded by using the Lorentzian line shapew ith a linew idth of 10 cm-1.coloronline
3.2 PyS-Agn簇结构的拉曼光谱(n=3,5,7,9)
对于4MPY吸附在银表面,我们首先考虑了PYSH异构体以巯基硫分别与四个小银簇的作用。4MPY可与银纳米粒子表面三种吸附位作用,即顶位、桥位和穴位。基于我们以前的计算23,在这里我们选择了PYSH与银簇Ag3、Ag5、Ag7和Ag9的桥位作用。在这些优化结构中,Ag―S键长约为0.2458-0.2644 nm。与PYSH的优化结构中C―S键长0.1778 nm相比,当PYSH与银簇作用后,吸附C―S键长约为0.1765-0.1794 nm。
图4是计算四种复合物优化结构的拉曼光谱。首先,当4MPY分子以巯基端与Ag簇形成复合物时,DFT计算标度频率约为990、1090和1576 cm-1。可以看到4MPY分子以巯基端与不同银簇作用时,银簇的大小对理论计算频率和谱峰相对强度影响不明显。这主要是由于巯基端与银簇形成很强的S―Ag键,这样强的化学吸附作用对吡啶环中的振动影响较小23。其次,当4MPY分子以巯基端与Ag离子形成复合物时,DFT计算标度频率分别为991、1092和1580 cm-1。实验测定4MPY银盐的这三个强的拉曼光谱峰依次为1007、1107和1586 cm-1,同时实验观测的SERS谱峰为1010、1098和1580 cm-1,均比理论值24,30,31大。对于PySAg复合物,界面环境效应,如溶剂化或质子化有必要进一步考虑。
3.3 质子化和溶剂化效应
上面我们看到4MPY与金属簇作用,其环振动频率比实验观测频率低,认为除巯基硫与银簇成键的影响外,有待进一步考虑溶剂水的氢键作用和质子化作用。在研究吡啶的拉曼光谱时,人们已注意到吡啶环v1振动模的频率依赖于弱的氢键作用,如吡啶分子气态时其振动频率为992 cm-1;在水溶液中,其频率蓝移到1008 cm-1;当与金属或金属离子作用,可以进一步蓝移到1020 cm-1以上32-34。当4MPY以巯基与银簇作用后,在下面我们比较了N端与水形成氢键作用以及质子化后对4MPY拉曼光谱的影响。
图5是计算Ag5-S-PYSN与水形成氢键以及吡啶氮质子化后的拉曼光谱。与图4的计算拉曼光谱相比,吡啶环呼吸振动的频率明显蓝移,但继续增加水分子数目,振动频率的位移不发生明显变化。当仅考虑一个水分子与吡啶环氮原子作用时,吡啶环v12振动频率蓝移到998.5 cm-1。进一步考虑增加水分子数目4和8,4MPY分子的v12振动模的频率保持在1000.0和999.5 cm-1。进一步利用SMD模型考虑长程的溶剂化效应,计算频率不发生明显变化,如v1和v8a模的振动谱峰仍位于1190和1580 cm-1。上述计算结果表明4MPY的吡啶氮与水形成氢键作用,使吡啶环呼吸振动频率蓝移,并与实验观测频率趋向一致。
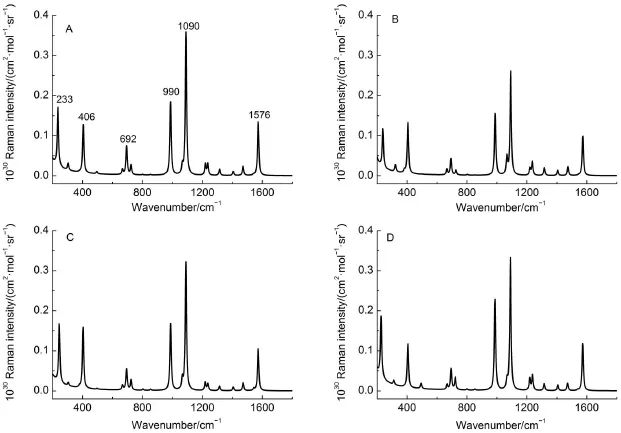
图4 B3LYP/6-311+G**/LANL2DZ水平计算PyS-Agn复合物的模拟拉曼光谱Fig.4 Simulated Raman spectra of PyS-Agncomp lexes calcu lated at the B3LYP/6-311+G**/LANL2DZ level(A)Ag3-S-PyN;(B)Ag5-S-PyN;(C)Ag7-S-PyN;(D)Ag9-S-PyN.The Raman intensity isestimated by differentialRaman scattering cross section. It isexpanded according to the Lorentzian line shapew ith the linew idth about10 cm-1.
图5 B考虑了单端作用4MPY的吡啶氮位质子化作用的影响14。当PyS-Ag5的吡啶环氮位质子化,其特征频率是988、1043、1587和1625 cm-1。当对质子化结构进行溶剂化后,水合质子溶剂化后的模拟拉曼光谱如图5B实线所示。对仅质子化结构溶剂化导致1043谱峰蓝移至1091 cm-1,其它三个谱峰为992、1578和1621 cm-1。当考虑对吡啶环氮位与水合质子复合结构溶剂化时,所得计算结果表明,1578 cm-1谱峰强度减小,特征谱峰为992、1086和1622 cm-1。另外,在这种情况下较强的谱峰还有415和1383 cm-1。
3.4 双端作用
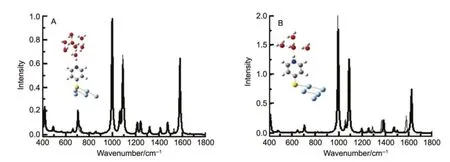
图5 B3LYP/6-311+G**/Lanl2DZ水平计算模拟拉曼光谱Fig.5 Simulated Raman spectra of comp lexes calculated at the B3LYP/6-311+G**/Lan l2DZ level(A)Ag5-SPYN-(H2O)8;(B)Ag5-SPYNH-(H2O)4.The excitationwavelength is632.8 nm.TheRaman intensity isestimated according to differentialRaman scattering cross sectionsexpanded in the Lorentzian line shapew ith the linew idth about10 cm-1.
有关4MPY以双端作用的SERS研究还很少22,23。下面我们考虑当巯基硫和吡啶环氮同时与银簇作用时,采用Ag5-S-PYSN-N-Ag4结构作为表面吸附构型,比较双端吸附对4MPY拉曼光谱的影响。这里吡啶环上氮端与Ag4簇作用,这种结构成键较强,其主要原因是Ag4簇的最低未占据轨道可以和吡啶氮上孤对电子形成强的配位键34。在Ag5-S-PYSN-N-Ag4结构中,Ag―S键长分别为0.2579和0.2576 nm,C―S键长是0.1782 nm。在氮端与银簇作用形成的N―Ag键长为0.2305 nm。
图6是计算双端优化结构的拉曼光谱。在气相情况下,从振动谱峰频率位置来看,DFT计算4MPY单端吸附的特征谱峰分别为991、1092和1580 cm-1。当4MPY以双端与银簇作用,如图6A所示,其特征谱峰为1010、1098和1592 cm-1,这与实验观测的谱峰1010、1098和1580 cm-1更为接近5。图6B是用SMD模型考虑水的溶剂化效应的拉曼光谱,特征频率为1003、1093和1584 cm-1。可以看到当考虑到吡啶环氮与银作用,计算频率与实验振动频率进一步趋于吻合。考虑吡啶环氮孤对与银作用时,不仅改进对1065 cm-1振动谱峰强度的理论预测,而且该峰的强度明显增强。最后,在以上的计算拉曼光谱中,有两个基频峰在1200-1300 cm-1区间,其分别对应于苯环上C―H对称(v9a)和反对称(v9b)面内弯曲振动,与前面讨论的谱峰相比,其拉曼强度明显较弱。但在实验数据中,在这一区域可以看到较强的SERS谱峰13,14。为此下面我们将进一步考虑电荷转移机理对SERS谱强度的影响。
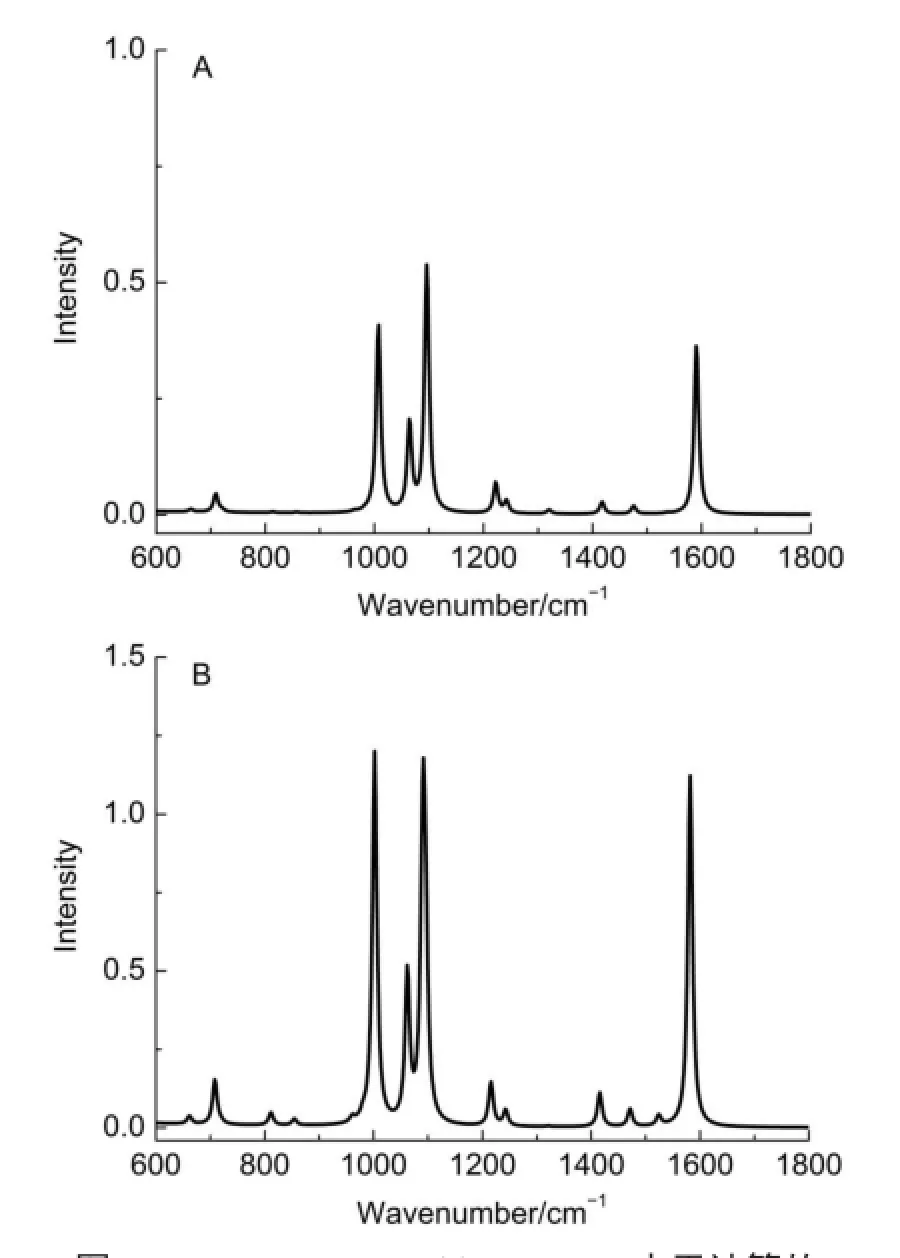
图6 B3LYP/6-311+G**/Lanl2DZ水平计算的双端吸附结构Ag4-N-PyS-Ag5的模拟拉曼光谱Fig.6 Simulated Raman spectra of Ag4-N-PyS-Ag5calcu lated at the B3LYP/6-311+G**/Lanl2DZ level(A)gas phase;(B)SMDmodel. The excitationwavelength is632.8 nm.
3.5 光驱电荷转移态
光驱电荷转移机理已被用于解释4MPY的SERS光谱13,20,21。文献13是基于实验观测结果,提出在拉曼激发光辐射下发生从银到4MPY的电荷转移。文献20考虑4MPY以巯基硫与Ag13簇的作用,提出电荷转移是从分子到银簇。他们考虑的电荷转移态跃迁能为1.96 eV,但是理论计算拉曼光谱与实验SERS光谱相差较大,特别是在低波数出现多个强峰,而在1000和1220 cm-1处的特征SERS谱峰却很弱。文献21分别考虑了4MPY以巯基硫和吡啶氮与Ag20簇作用,但她们的计算从银簇到分子的电荷转移态能量均大于3.1 eV,远大于实验中所用的激光能量。所以,目前对4MPY吸附在银表面,其电荷转移增强机理存在两个问题:一是4MPY吸附结构与电荷转移态的关系;二是电荷转移增强哪些4MPY的活性振动模。基于以上问题,我们考虑了两种4MPY吸附模型,即PYSN-S-Ag5和Ag5-S-PYSN-N-Ag4。由于在SERS实验中,所用的激光能量要小于金属的带间跃迁能,即仅涉及金属价层sp带的电子跃迁。对于银,我们主要考虑银簇的5s轨道与4MPY的前线轨道作用,以及相关的电子跃迁性质。
图7是用两种模型复合物计算的考虑低能激发态的拉曼光谱。对于Py-S-Ag5复合物,TD-B3LYP计算第一激发态(S1)和第二激发态(S2)的E1=2.64 eV和E2=3.04 eV,其振子强度分别为f1=0.0069和f2=0.3228。如果用SMD模型考虑溶剂化效应,则得到E1=2.50 eV,f1=0.0031;E2=2.91 eV,f2=0.8403。在不考虑溶剂化效应情况下,当采用632.8 nm激发光波长时,基于耦合微扰方法计算预共振拉曼光谱的理论方法,除绝对强度上增强外,Py-S-Ag5簇的计算拉曼光谱与静态的拉曼光谱类似。当考虑采用488.5 nm激发波长,其能量比S1态的低0.1 eV,计算拉曼光谱如图7A所示。在计算拉曼光谱中,展示强拉曼活性的是420和1110cm-1谱峰。它们分别是吡啶环的v6a弯曲振动和v1环呼吸振动,二者对C―S键和Ag―S键更为敏感。
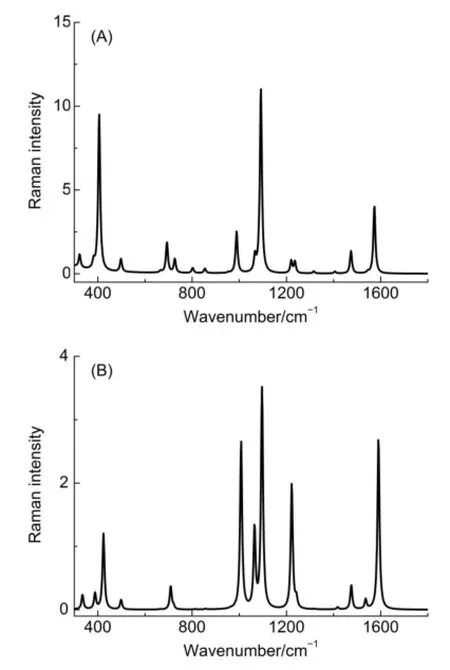
图7 基于B3LYP/6-311+G**/LANL2DZ方法和拉曼光谱理论计算(A)在488.5 nm的单端构型Py-S-Ag5和(B)在550.0 nm的双端结构Ag4-N-PyS-Ag5的频率相关拉曼光谱Fig.7 Frequency-dependen t Raman spectra of 4M PY w ith a silver cluster in(A)a single-end configuration Py-S-Ag5at488.5 nm and(B)a two-end configuration Ag5-S-PYSN-N-Ag4at550.0 nm calcu lated by combining the B3LYP/6-311+G**/LANL2DZmethod and Ram an intensity theoryThe Raman spectraare drawn by using differential Raman scattering cross section according to the Lorentzian lineshapew ith the linew idth about10 cm-1.
在双端情况下,采用TD-B3LYP计算Ag5-SPy-N-Ag4低能激发态,除S8态来自Ag5簇内部跃迁外,能量低于3.0 eV有10个单重态,均是由吡啶环成键Ag4簇激发所引起的电子跃迁,它们包括了Ag4簇内部电子激发态(S10),Ag4簇到吡啶环的电荷转移激发态(S3、S6和S7)以及Ag4到Ag5之间的电荷转移态(S1、S2、S4、S5、S8和S11)。对于3个电荷转移态S3、S6和S7,它们的跃迁能和振子强度分别是E3=1.50 eV,f3<0.0001;E6=2.09 eV,f6=0.0001;和E7=2.39 eV,f7=0.0006。它们的跃迁振子强度均很小,这暗示它们对光的吸收几率非常小,但其显著地影响拉曼散射过程,对SERS相对强度有显著的影响。图7B是我们采用激发波长为550 nm时计算拉曼光谱。该计算拉曼光谱很好地解释了当考虑吡啶环氮与银簇作用,导致增大位于1220 cm-1的v9a模拉曼强度。除v12、v1和v8a振动谱峰明显增强外,因电荷转移态的共振增强效应,其显著增强吡啶环面内弯曲振动v9a模的拉曼强度。
4 结论
结合DFTB3LYP理论计算和拉曼光谱理论对4MPY分子进行了研究,构建了4MPY吸附结构与拉曼光谱之间的对应关系。计算结果解释了在气相、极性溶剂水和固体粉末中4MPY异构体的相对稳定性。在后两种情况下4MPY发生异构化,主要以两性离子形式存在,且4MPY分子之间或与溶剂分子之间形成氢键作用,影响4MPY分子的拉曼光谱。在纳米结构银或粗糙银表面,与4MPY仅以巯基硫的单端吸附构型相比,我们提出了4MPY存在双端吸附构型。
当4MPY以双端吸附构型与表面作用,其吡啶环氮端与银作用产生两种显著变化。一方面会导致吡啶环三角弯曲振动v12蓝移。另一方面,相对于4MPY以巯基硫与银作用的单端吸附构型,双端吸附构型的电荷转移态具有低跃迁能量,这与目前大多数SERS实验所采用的激发波长相近。基于4MPY的双端吸附结构,计算的拉曼光谱很好地解释了实验观测吡啶环上v9a对称C―H面内弯曲振动峰。我们的理论计算结果表明,v9a振动模的强度变化与吡啶氮端与银作用时发生电荷转移密切相关。因此,实验观测这个谱峰显著增强,暗示电荷转移增强机理对SERS化学增强效应有贡献。
类似于苯硫酚在金属表面吸附,4MPY以巯基硫吸附在金属表面有大的倾斜角,这有利于吡啶环氮与纳米结构金属表面再作用。由于纳米结构表面的复杂性,两种吸附结构可能同时存在,实验测得的SERS来自表面不同吸附构型的总贡献。最后,我们工作可能拓展4MPY分子在其他金属表面的SERS研究,如具有强SPR效应的金和铜纳米结构表面。由于金和铜的d带更靠近其Ferm i能级,吡啶环氮原子与金和铜的成键作用更强。因此,4MPY在这两种金属表面以巯基硫成键时,也有利于其吡啶环氮与金和铜表面发生吸附作用。
Re ferences
(1)W ilson,E.B.;Decius,J.C.;Cross,P.C.Molecular Vibrations: The Theory of Infrared and Raman Vibrational Spectra;Dover: New York,1955.
(2)Wang,Z.J.;Rothberg,L.J.J.Phys.Chem.B 2005,109,3387. doi:10.1021/jp0460947
(3)W u,D.Y.;Li,J.F.;Ren,B.;Tian,Z.Q.Chem.Soc.Rev.2008, 37,1025.doi:10.1039/B707872M
(4)W u,D.Y.;Zhang,M.;Zhao,L.B.;Huang,Y.F.;Ren,B.;Tian, Z.Q.Sci.China-Chem.2015,58,574.doi:10.1007/s11426-015-5316-y
(5)Wu,D.Y.;Liu,X.M.;Duan,S.;Xu,X.;Ren,B.;Lin,S.H.; Tian,Z.Q.J.Phys.Chem.C 2008,112,4195.doi:10.1021/ jp0760962
(6)Su,S.;Huang,R.;Zhao,L.B.;Wu,D.Y.;Tian,Z.Q.Acta Phys.-Chim.Sin.2011,27,781.[苏抒,黄荣,赵刘斌,吴德印,田中群.物理化学学报,2011,27,781.]doi:10.3866/PKU. WHXB20110418
(7)Gui,J.Y.;Lu,F.;Stern,D.A.;Hubbard,A.T.J.Electroanal. Chem.1990,292,245.doi:10.1016/0022-0728(90)87339-L
(8)Hu,J.W.;Zhao,B.;Xu,W.Q.;Li,B.F.;Fan,Y.G. Spectrochimica Acta A 2002,58,2827.doi:10.1016/S1386-1425 (02)00074-4
(9)Singh,P.;Deckert,V.Chem.Commun.2014,50,11204.doi: 10.1039/C4CC04642K
(10)Latorre,F.;Kupfer,S.;Bocklitz,T.;Kinzel,D.;Trautmann,S.; Graefe,S.;Deckert,V.Nanoscale 2016,8,10229.doi:10.1039/ C6NR00093B
(11)Carron,K.T.;Hurley,L.G.J.Phys.Chem.1991,95,9979.doi: 10.1021/j100177a068.
(12)Bron,M.;Holze,R.J.So lid State Electrochem.2015,19,2673. doi:10.1007/s10008-015-2869-9
(13)Shegai,T.;Vaskevich,A.;Rubinstein,I.;Haran,G.J.Am. Chem.Soc.2009,131,14390.doi:10.1021/ja904480r
(14)Chao,Y.W.;Zhou,Q.;Li,Y.;Yan,Y.R.;Wu,Y.;Zheng,J.W. J.Phys.Chem.C 2007,111,16990.doi:10.1021/jp0760051
(15)Zheng,X.S.;Hu,P.;Zhong,J.H.;Zong,C.;Wang,X.;Liu,B. J.;Ren,B.J.Phys.Chem.C 2014,118,3750.doi:10.1021/ jp409711r
(16)Yu,H.Z.;Xia,N.;Liu,Z.F.Anal.Chem.1999,71,1354.doi: 10.1021/ac981131+.
(17)Wang,Y.;Yu,Z.;Ji,W.;Tanaka,Y.;Sui,H.;Zhao,B.;Ozaki,Y. Angew.Chem.-Int.Edit.2014,53,13866.doi:10.1002/ anie.201407642
(18)Respondek,I.;Benoit,D.M.J.Chem.Phys.2009,131,054109. doi:10.1063/1.3193708
(19)Sun,L.;Bai,F.Q.;Zhang,H.X.Acta Phys.-Chim.Sin.2011, 27,1335.[孙磊,白福全,张红星.物理化学学报,2011,27, 1335.]doi:10.3866/PKU.WHXB20110602
(20)Birke,R.L.;Lombardi,J.R.J.Optics 2015,17,114004.doi: 10.1088/2040-8978/17/11/114004
(21)Liu,L.;Chen,D.;M a,H.;Liang,W.J.Phys.Chem.C 2015, 119,27609.doi:10.1021/acs.jpcc.5b05910
(22)Iida,K.;Noda,M.;Nobusada,K.J.Chem.Phys.2014,141, 124124.doi:10.1063/1.4896537
(23)Wu,D.Y.;Liu,X.M.;Huang,Y.F.;Ren,B.;Xu,X.;Tian,Z.Q. J.Phys.Chem.C 2009,113,18212.doi:10.1021/jp9050929
(24)Jung,H.S.;Kim,K.;Kim,M.S.J.Mol.Struct.1997,407,139. doi:10.1016/S0022-2860(97)00006-9
(25)Guo,H.;Ding,L.;Mo,Y.J.J.Mol.Struct.2011,991,103.doi: 10.1016/j.molstruc.2011.02.012
(26)Zhang,L.;Bai,Y.;Shang,Z.G.;Zhang,Y.K.;M o,Y.J. J.Raman Spectrosc.2007,38,1106.doi:10.1002/jrs.1719
(27)Etter,M.C.;Macdonald,J.C.;Wanke,R.A.J.Phys.Org. Chem.1992,5,191.doi:10.1002/poc.610050404
(28)Flakus,H.T.;Tyl,A.;Jones,P.G.Spectroc.Acta Pt.A-Molec. Biomo lec.Spectr.2002,58,299.doi:10.1016/S1386-1425(01) 00526-1
(29)M uthu,S.;Vittal,J.J.Cryst.Grow th Des.2004,4,1181.doi: 10.1021/cg0498812
(30)Baldw in,J.;Schuhler,N.;Butler,I.S.;Andrews,M.P. Langmuir1996,12,6389.doi:10.1021/la960367a
(31)Baldw in,J.A.;Vlckova,B.;Andrews,M.P.;Butler,I.S. Langmuir 1997,13,3744.doi:10.1021/la960719d
(32)Schlucker,S.;Singh,R.K.;Asthana,B.P.;Popp,J.;Kiefer,W. J.Phys.Chem.A 2001,105,9983.doi:10.1021/jp0122272
(33)Wu,D.Y.;Ren,B.;Jiang,Y.X.;Xu,X.;Tian,Z.Q.J.Phys. Chem.A 2002,106,9042.doi:10.1021/jp025970i
(34)Wu,D.Y.;Hayashi,M.;Shiu,Y.J.;Liang,K.K.;Chang,C.H.; Yeh,Y.L.;Lin,S.H.J.Phys.Chem.A 2003,107,9658.doi: 10.1021/jp034951l
Density Functional Theoretical Study on SERS Chemical Enhancement Mechanism of 4-Mercaptopyridine Adsorbed on Silver
WU Yuan-Fei LIMing-Xue ZHOU Jian-Zhang WU De-Yin*TIAN Zhong-Qun*
(DepartmentofChemistry,College ofChemistry and ChemicalEngineering,State Key Laboratory ofPhysicalChemistry of Solid Surfacesand Collaborative Innovation Centerfor EnergyMaterialsChemistry,Xiamen University,Xiamen 361005, Fujian Province,P.R.China)
Su rface-enhanced Ram an spec troscopy(SERS)is one of the m ost powe rfu l techniques for obtaining fingerprint information onmolecules adsorbed on coinagemetalsurfaces.Its detection sensitivity has reached the single-molecule level.On the basis ofdensity functiona l theoretical(DFT)calculations and Raman scattering theory,we investigated the normalRaman spectra of two isomers and surface-enhanced Raman scattering(SERS)spectra of4-mercaptopyridine(4MPY)adsorbed on silver.The results aided us in uncovering the relationships be tween norma l Raman spectra and SERS spec tra and adso rp tion con figura tion, tautomerization,protonation,and hyd rogen bonding interactions as wellas low-lying excited states.First,we compared the relative stability and normalRaman spectra of two isomers of4MPY in the gas phase and aqueous solution with a solventmodelsim ilar to the solvationmodelofdensity(SMD).We then studied the Raman spectra of4MPY interacting with silver clusters.Our results indicate that the Raman spectra were notdependenton the size of the silver clusters,ow ing to the forma tion of strong Ag―S bonds.W e also conside red two cases ofN-end interaction in the 4MPY-Ag5complex.(1)For the hydrogen bond interaction between the nitrogen in 4MPY and water c lusters o r hydrated proton clusters,the theoretical results indicated that the vibrational frequencies of the pyridine ring increase.(2)For the interaction of the 4MPY-Ag5comp lexw ith a silver c lusterAg4through the lone-paired orbitalin nitrogen of the pyridine ring,the theoretical results further revealed that the vibrational frequency shift is in good agreementw ith SERS peaks reported in the literature.Finally,our calcu lated results focused on the relationship between the Raman spectra and the charge transfermechanism when the excitation photonic ene rgy m atches the transition energy of low-lying excited states in single-end and doub le-end adsorption configuration.Particularly for the case of the double-end adsorption configuration,the charge transfer states from the excitation from the silverclusterbinding to the pyridine ring notonly enhance the Raman signals of v12,v1,and v8am odes,but also selectively enhance the Raman signalof the v9am ode associated w ith the symmetric C―H in-plane bending vibration.
Surface-enhanced Raman spectroscopy;Density functional theory;Charge transfer mechanism;4-Mercaptopyridine;Silver
O646
10.3866/PKU.WHXB201611211
www.whxb.pku.edu.cn
Received:August2,2016;Revised:November21,2016;Published online:November21,2016.
*Corresponding authors.WU De-Yin,Email:dywu@xmu.edu.cn;Tel:+86-592-2189023.TIAN Zhong-Qun,Email:zqtian@xmu.edu.cn; Tel:+86-592-2186979.
The projectwassupported by the NationalNaturalScience Foundation of China(21273182,21533006,21373172).
国家自然科学基金(21273182,21533006,21373172)资助项目©Editorialoffice of Acta Physico-Chim ica Sinica
