蟾毒配基脂质-蛋白杂化纳米粒的制备及评价
2017-01-05李晚秋
李晚秋,林 霞,许 航,唐 星*
(1. 沈阳药科大学 中药学院,辽宁 沈阳 110016; 2. 上海交通大学 药学院,上海 200240;3. 沈阳药科大学 药学院,辽宁 沈阳 110016)
蟾毒配基脂质-蛋白杂化纳米粒的制备及评价
李晚秋1,林 霞2,许 航3,唐 星3*
(1. 沈阳药科大学 中药学院,辽宁 沈阳 110016; 2. 上海交通大学 药学院,上海 200240;3. 沈阳药科大学 药学院,辽宁 沈阳 110016)
目的采用搅拌超声法制备蟾毒配基脂质-蛋白杂化纳米粒,延缓药物的体外释放。方法以单因素考察法优化处方及制备工艺,并应用透射电镜观察制剂形态,动态光散射法测定粒径和 ζ-电位,差示扫描量热法和 X-射线衍射法表征药物在制剂中的存在形式,透析法考察纳米粒及溶液剂中药物的释放行为。结果纳米粒外观圆整均匀,平均粒径为(82.4 ± 28.5) nm,ζ-电位为-19.44 mV,药物在制剂中以无定形态存在,与溶液剂相比,纳米粒可显著延缓药物释放,其释放行为符合Weibull模型,释放机制以扩散为主。结论利用搅拌超声法,以白蛋白和卵磷脂作为载体和稳定剂制备的脂质-蛋白杂化纳米粒可显著延缓蟾毒配基的体外释放。
药剂学;脂质-蛋白杂化纳米粒;搅拌超声法;蟾毒配基;处方优化;制备工艺;体外释放
蟾酥为蟾 蜍 科 动 物 中 华 大 蟾 蜍( Bufo bufo gargarizans Cantor.)或黑眶 蟾 蜍(Bufo melanostictus Schneide)的耳后腺及 皮 肤腺分泌 的 白色浆液 经加工干燥而成[1-2],在临床上表现出显著的抗肿瘤活性,其中发挥抗肿瘤作用的主要成分是脂溶性的蟾毒配基(bufadienolides)化合物,蟾毒灵(bufalin, B)、华蟾酥毒基(cinobufagin, C)和脂蟾毒配基(resibufogenin, R)是蟾毒配基中含量较高、活性较强的三种主要有效成分,其化学结构式见图1。研究表明,蟾毒配基B、C和R对消化系统肿瘤显示出极强的细胞毒性,同时还具有镇痛和增强免疫力的作用,可改善病人生存质量,因此其在抗肿瘤方面显示出巨大潜力。但目前上市的蟾酥制剂有效成分纯度低,临床需要剂量大,患者顺应性差,且给药后在体内分布广泛,消除迅速,可引起严重的心脏毒性和神经毒性,使其应用受限。因此有必要开发一种作用持久、高效、低毒的蟾毒配基新剂型,以降低毒性及不良反应,充分发挥其抗肿瘤活性。

Fig. 1 Chemical structures of bufalin (B), cinobufagin (C) and resibufogenin (R)图1 蟾毒灵(B)、华蟾酥毒基(C)和脂蟾毒配基(R)的化学结构式
近年来,一种新型纳米给药系统正逐渐引起人们的关注,即脂蛋白纳米粒(lipoprotein-based nanoparticle)。脂蛋白具有天然球形结构,其中甘油三酯与胆固醇酯构成疏水核心,外层被磷脂及载脂蛋白包裹。脂蛋白作为药物载体有如下优势:(1)生物相容性好,在血液中稳定;(2)其疏水核心可包载一定量亲脂性药物,提高其溶解度和稳定性;(3)一些肿瘤细胞过度表达低密度脂蛋白受体(LDLR),这为脂蛋白纳米粒靶向肿瘤组织提供了可能性[3-4]。但是脂蛋白难以从人血清中大量提取[5],目前全合成的脂蛋白也无法用作纳米载体,且脂蛋白纳米粒难以用于高脂血症患者,使其应用受限。
受脂蛋白结构的启发,本研究以药物作为疏水核心,以卵磷脂和白蛋白作为载体和乳化剂,制备了蟾毒配基脂质-蛋白杂化纳米粒(bufadienolides-loaded lipid protein hybridization nanoparticles, BU-LP-NPs),拟通过肿瘤细胞过度表达LDLR的特性及肿瘤组织 本身特 有的EPR效 应,实现药物的肿瘤靶向。本文采用搅拌超声法制备了BU-LP-NPs,并通过单因素考察法优化了处方和制备工艺,最后对制剂的理化性质及体外释放进行评价,初步解决蟾毒配基半衰期短、消除迅速的问题。
1 仪器与材料
超声细胞破碎仪(Sonics & Meterials. INC.,USA),NicompTMPSS 380动态光散射粒度测定仪(Santa Barbara, USA),DF-101S集热式恒温磁力搅拌器(巩义英峪予华仪器厂),VirTis冻干机(SP Industries, NY,USA),N-1001旋转蒸发仪(东京理化器械株式会社),ATL-124电子分析天平、PB-10精密pH计(赛多利斯科学仪器有限公司),Chromaster 5000高效液相色谱仪(日本日立公司),Amicon Ultra-0.5离心超滤管(美国millpore公司),雷勃尔LG16-C离心机(北京雷勃尔离心机有限公司),JEM-2100透射电镜(JEOL, Tokyo, Japan),DSC-60差热分析仪、TGA 50热重分析仪(Shimadzu Co., Kyoto, Japan),D/Max 2400 X-射线粉末衍射仪(日本理光公司),透析袋(截留分子质量1.4×104u,上海绿鸟科技发展有限公司)。
蟾毒配基(自制,以主要成份B、C和R含量之和计,其纯度质量分数>99%,批号 20130118),人血白蛋白(兰州生物制品研究所有限责任公司),注射用蛋黄卵磷脂PL-100M、PC-98T(上海艾维特公司),注射用蛋黄卵磷脂Lipoid E80®、Lipoid S-100(德国Lipoid®公司),大豆卵磷脂(上海太伟药业有限公司),蔗糖、乳糖、葡萄糖、甘露醇、麦芽糖(天津博迪化工有限公司),海藻糖(南宁中诺生物工程有限责任公司),吐温-80(上海申宇医药化工有限公司),乙腈、甲醇(色谱纯,天津康科德试剂公司),其他试剂(分析纯,市售)。
2 方法与结果
2.1 纳米粒的制备
采用搅拌超声法制备BU-LP-NPs。精密称取蟾毒配基20 mg和蛋黄卵磷脂Lipoid E80 200 mg,置于西林瓶中,加入无水乙醇4 mL,超声混匀,作为油相;配制pH值6.5的磷酸盐缓冲液10 mL,加热至60 ℃,作为水相。于400 r·min-1磁力搅拌下,将油相缓缓滴入水相中,滴完后继续搅拌10 min,旋蒸除去乙醇,向得到的混悬液中加入白蛋白溶液1.0 mL,冰水浴中探头超声3 min(300 W,超声3 s,间歇1 s),将混悬液用0.22 μm微孔滤膜过滤除菌,分装,冻干,即得。
2.2 产率及包封率的测定
2.2.1 色谱条件
色谱柱:HiQsil C18柱(250 mm × 4.6 mm, 5 µm);流动相:乙腈-5 g·L-1磷酸二氢钾溶液(体积比45∶55,用磷酸调节水相pH值为3.2);检测波长:296 nm;流速:1.0 mL·min-1;柱温:40 ℃;进样量:10 µL。
2.2.2 产率的测定
精密量取过滤后的BU-LP-NPs 1.0 mL,置于50 mL量瓶中,加适量甲醇超声溶解并定容,摇匀,滤过。取续滤液,以HPLC法进样分析,测得过膜后制剂中3个蟾毒配基的含量之和(Qn),投药量计为Q,则产率(Yield, %)= Qn/Q × 100%。
2.2.3 包封率的测定
包封率(Entrapment efficiency, EE)是制剂处方工艺筛选及质量评价的重要指标,且对制剂的稳定性具有重要影响,因此本研究采用离心超滤法测定BU-LP-NPs的包封率。具体操作如下:
药物总含量的测定: 精密量取BU-LP-NPs溶液1.0 mL,置于50 mL量瓶中,加甲醇超声溶解并定容,摇匀,滤过。取续滤液,用HPLC法测定纳米粒中3个蟾毒配基的质量浓度。
水相中药物含量的测定: 精密量取BU-LP-NPs溶液0.5 mL,置于超滤管中,然后放在配套接收管内,4 000 r·min-1离心20 min,取接收管内的滤液进样分析,测定水相中3个蟾毒配基的质量浓度。
其中:ρtotal为制剂中蟾毒配基总质量浓度,Vtotal为制剂总体积,ρwater为水相中蟾毒配基的质量浓度,Vwater为水相体积。
由于纳米粒制备过程中,有机相最终被除去,因此Vwater≈Vtotal,包封率计算公式可简化为:。
2.3 制备工艺的考察
2.3.1 制备工艺的选择
为了制备粒径较小、产率较高的BU-LP-NPs,作者主要比较了薄膜分散法和搅拌超声法两种制备工艺。工艺研究阶段所用磷脂为Lipoid E80,固定其用量为20 g·L-1。
薄膜分散法:将处方量的蟾毒配基、卵磷脂和乙醇混合,超声使溶解,作为油相。将油相置于茄形瓶中,旋蒸除去乙醇,得到均匀的磷脂薄膜。向茄形瓶中加入适量蒸馏水,水化30 min,向混悬液中加入人血清白蛋白(HSA),探头超声3 min,即得。
搅拌超声法:取处方量的药物、卵磷脂和乙醇,超声混匀,作为油相。磁力搅拌下将油相缓缓滴入磷酸盐缓冲液中,继续搅拌10 min,旋蒸除去乙醇,向得到的混悬液中加入白蛋白,冰水浴中探头超声3 min,即得。
结果表明,采用薄膜分散法制备BU-LP-Nps时,形成的磷脂薄膜不均匀,水化后仍有少量磷脂复合物团块存在,即使超声也难以使其分散均匀,使制得的纳米粒中存在大粒子,粒度分布很宽,且重现性差;而搅拌超声法制备的BU-LP-Nps粒径小且分布均匀,产率较高,稳定性好,其操作过程较薄膜分散法简便,因此本研究采用搅拌超声法制备BU-LP-Nps。
2.3.2 制备温度的考察
磷脂存在相转化温度(phase inversion temperature, θPI),在此温度下,磷脂聚集在油/水界面的倾向较大,乳化能力相对较强[6],因此在相转化温度附近(约70 ℃)制备纳米粒时粒径较小,但温度过高不利于磷脂及药物的稳定性,因此将制备温度控制在 60 ℃,旋蒸除去乙醇后迅速冷却,再加入HSA探头超声,可制得粒径较小且稳定的纳米粒。
2.3.3 油相滴入速度的考察
分别将固定体积的油相以10、1.0及0.4 mL·min-1的速度用注射器加入水相中,其他制备条件固定,制备BU-LP-Nps。结果发现:滴加速度为10 mL·min-1时,制得的纳米粒粒度分布宽,且有大粒子存在,可能是由于短时间内加入大量油相,形成较多新生界面,乳化剂与界面结合不充分,导致粒子聚集,产生大粒子;滴加速度为1.0及0.4 mL·min-1时,形成的纳米粒粒径相近(0.4 mL·min-1滴入时粒度分布稍宽),为减少药物及磷脂的受热时间,将油相滴入速度控制在1.0 mL·min-1。
2.3.4 搅拌速度的考察
将油相加入水相后,需以一定的速度搅拌,使两相充分乳化。设定搅拌速度分别为200、 300、400和500 r·min-1,其他条件固定,制备BU-LP-Nps,分别测定其粒径,结果见图2。结果表明,随着搅拌速度增大,粒径及粒度分布均变小,当搅拌速度为500 r·min-1时,粒径降低不明显,且搅拌速度过大易引起液体溅出,影响乳化效果及产率,因此,将搅拌速度控制为400 r·min-1。
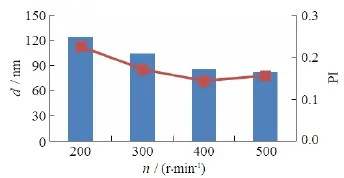
Fig. 2 The effect of stirring speed on the PSD of BU-LP-NPs.图2 搅拌速度对蟾毒配基脂质-蛋白杂化纳米粒粒径的影响
2.3.5 搅拌时间的考察
形成初乳后应搅拌一定的时间,使两相充分分散。但高温条件下搅拌时间过长,会造成磷脂氧化、水解,且不利于药物的稳定,因此搅拌时间不宜过长。油相滴加完毕后,分别继续搅拌10、15 和20 min,结果表明,搅拌时间对粒径影响不大。因此,将搅拌时间确定为10 min。
2.3.6 超声时间及功率的考察
固定超声功率及其他制备条件,考察不同超声时间对BU-LP-Nps粒径的影响。分别将超声时间设为3、5、7 和9 min,结果发现随着超声时间延长,粒径及粒度分布均变大,超声3 min的样品4 ℃下放置3 d稳定性良好,而超声7和9 min的样品4 ℃放置,1 d内即有药物析出,因此将超声时间定为3 min。固定超声时间,增大超声功率,粒径有降低的趋势,但超声功率在300 W以上时,粒径降低不明显且粒度分布变宽,而超声功率过大对仪器的磨损也较大,因此确定超声功率为300 W(超声3 s,间歇1 s)。
2.4 处方筛选
2.4.1 磷脂种类的选择
磷脂是人体的内源性物质,与生物膜结构相似,具有良好的生物相容性,因此是脂质-蛋白杂化纳米粒的首选乳化剂[7]。固定磷脂及白蛋白的质量浓度为20 g·L-1,其他制备条件不变,分别向油相中加入蛋黄卵磷脂PL-100M、Lipoid S-100、Lipoid E80、PC-98T及大豆卵磷脂(Lipoid TW),按“2.1”条方法制备BU-LP-NPs,考察磷脂种类对制剂外观及粒径的影响,结果见表1。
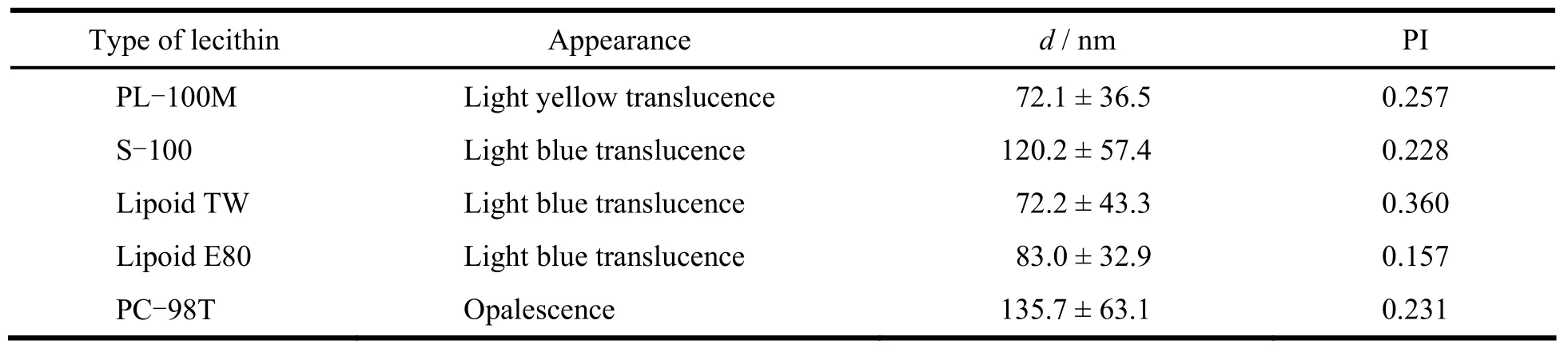
Table 1 The effect of lecithin type on the appearance and PSD of BU-LP-NPs表1 磷脂种类对蟾毒配基脂质-蛋白杂化纳米粒外观和粒径的影响
由表1可见,除PC-98T外,用其余磷脂制备的BU-LP-NPs外观均良好,粒径均较小,但只有用Lipoid E80时粒度分布较窄。为了使BU-LP-NPs长期稳定储存,最终需要将其冻干,因此在不加冻干保护剂的条件下,将用各种磷脂制备的BU-LP-NPs冷冻干燥,考察所选磷脂的冻干稳定性,结果见表2。结果表明,磷脂种类为Lipoid E80时,冻干产品外观较好,冻干前后粒径变化最小且复溶后透光性相对较好,因此将磷脂种类确定为Lipoid E80。

Table 2 The appearance and particle size of BU-LP-NPs containing different types of lecithin after lyophilization without cryoprotectant表2 含不同种类磷脂的蟾毒配基脂质蛋白杂化纳米粒(不加冻干保护剂)冻干后的外观和粒径
2.4.2 药脂质量比的考察
固定投药量及其他制备条件,分别加入不同比例的Lipoid E80,使药脂质量比分别为1∶5、 1∶8和1∶10,制备BU-LP-NPs,考察不同药脂质量比对制剂粒径及包封率的影响,结果见表3。结果表明,随着药脂质量比增大,粒径及粒度分布均变小,3种蟾毒配基的包封率也随之增大,且实验发现药脂质量比为1∶8时制得的样品室温放置12 h后,有药物析出,而药脂比为1∶10时,所制样品在室温下放置3 d后未见药物析出,且粒径增大不明显,因此将药脂质量比确定为1∶10。

Table 3 The effect of drug-lecithin ratio on the PSD and EE of BU-LP-NPs (n=3)表3 药脂比对蟾毒配基脂质-蛋白杂化纳米粒粒径和包封率的影响(n=3)
2.4.3 白蛋白质量浓度的选择
固定药脂质量比为1∶10,油水相体积比为1∶2.5,其他制备条件不变,分别在水相中加入不同体积的HSA(200 g·L-1),使其质量浓度分别为0、5、10和20 g·L-1,制备BU-LP-NPs,考察白蛋白质量浓度对制剂粒径及产率的影响,结果见表4。由表4可见,随着白蛋白质量浓度增大,纳米粒的粒径及粒度分布均降低,产率随之升高,当HSA质量浓度为20 g·L-1时,平均粒径为(85.9 ± 32.6) nm(<100 nm),产率可达到99.2%。而不加HSA时,将制剂在透射电镜下观察,粒子呈不规则球形,粒度分布不均匀且有聚集,难以形成纳米粒。因此,白蛋白对BU-LP-NPs具有一定的空间稳定作用,将形成的初乳探头超声时,大乳滴被分散成更多的小乳滴,加入适量白蛋白可稳定新生成的界面,使粒子不易聚集,从而使粒度分布均匀,有利于制剂的长期稳定,且白蛋白本身的表面活性作用可进一步降低粒径,从而提高BU-LP-NPs的产率。结果表明,HSA的质量浓度为20 g·L-1时,纳米粒的粒径和产率均较理想,因此确定白蛋白的质量浓度为2.0 g·L-1。

Table 4 The effect of HSA concentration on the PSD and yield of BU-LP-NPs (n=3)表 4 白蛋白质量浓度对蟾毒配基脂质-蛋白杂化纳米粒粒径和产率的影响(n=3)
2.4.4 油水比例的考察
固定药脂质量比为1∶10,白蛋白质量浓度为20 g·L-1,分别取不同体积的无水乙醇与药物、Lipoid E80混合,作为油相,使油相与水相体积比分别为1∶1.5、1∶2、1∶2.5和1∶10,其他条件不变,制备 BU-HSA-NPs,考察不同油水比例对制剂粒径的影响,结果见图 3。结果表明,油相比例过大或过小时,制剂粒径均较大。若油相比例太大(1∶1.5),滴入速度不变时,则油相加入时间过长,使得各粒子乳化时间不同,导致粒度分布不均匀,使粒子易于聚集,则制剂粒径较大;若油相比例过小(1∶10),则投药量一定时,油相中药物浓度过高,使得单个乳滴中所含药物较多,旋蒸除去乙醇时,单个乳滴析出的药物也较多,从而使粒径增大。由图3可见,当油水比例为1∶2时,制剂粒径最小,但粒度分布很宽,而为1∶2.5时,粒径及粒度分布均较小,因此将油相与水相的体积比确定为1∶2.5。

Fig. 3 The effect of oil-aqueous phase volume ratio on the PSD of BU-LP-NPs.图3 油水相体积比对蟾毒配基脂质-蛋白杂化纳米粒粒径的影响
2.4.5 水相pH的考察
在预实验中证实了pH值为6.0~8.0时,可制得较为稳定的BU-LP-NPs,其粒径随pH值增大而降低,pH值为8.0时粒径最小,pH值为6.5与7.0时粒径相差不大,但蟾毒配基在碱性条件下不稳定,因此不考虑碱性范围。此外,文献报道,大豆卵磷脂和蛋黄卵磷脂的水解受pH值影响,在pH 值6.5的缓冲液中其水解速率常数最小,从而使体系的pH值变化较小,进一步抑制了磷脂的水解[8]。综合考虑,选择pH值6.5的PBS缓冲液作为水相。
2.5 冷冻干燥工艺的考察
2.5.1 冻干工艺的考察
BU-LP-NPs在贮存期间易出现粒子聚集、药物渗漏等现象,且磷脂易氧化水解,使制剂稳定性降低,导致应用受限。采用冷冻干燥工艺将BU-LP-NPs固化,可有效解决这一问题,从而提高制剂的长期稳定性,采用的冻干曲线如图4所示。
2.5.2 冻干保护剂的筛选
冷冻干燥过程不利于脂质-蛋白杂化纳米粒结构的稳定,因为冻干过程中渗透压的改变、冰晶的形成、相转变及相分离等因素均会破坏磷脂的双分子膜,使其折叠、融合、破裂,导致药物渗漏[7],因此在冻干前应加入冻干保护剂,降低冷冻干燥对制剂的破坏作用,使纳米粒复溶后的粒径和形态无显著变化。常用的冻干保护剂有单糖、二糖和多元醇等,本研究考察了蔗糖、海藻糖、乳糖、麦芽糖、葡萄糖、氯化钠及甘露醇的冻干保护效果,比较冻干前后的粒径变化、冻干制剂的外观及水化后的透光性,以确定最佳冻干保护剂的种类和用量。
具体操作如下:取BU-LP-NPs混悬液2 mL8份,分别置于10 mL西林瓶中,分别加入上述冻干保护剂适量,乳糖的质量浓度为50 g·L-1,其余的冻干保护剂质量浓度为100 g·L-1,其中一份不加冻干保护剂,将样品溶液置于冻干机中,按上述工艺冷冻干燥。以冻干制剂的外观、复溶后的粒径、透光性及水化时间为评价指标,考察各冻干保护剂的效果,结果见表5。
结果表明:未加冻干保护剂以及加入氯化钠、葡萄糖冻干后的制剂外观萎缩坍塌,复溶时间均超过90 s,且复溶后可见大量固体物质,说明BU-LP-NPs的微观结构已经被破坏,粒子之间发生聚集,产生了脂质团块;以乳糖作为保护剂,冻干后的样品外观较好,有少量鳞状结晶,但复溶后溶液呈乳浊状,复溶时间长且透光性差;以甘露醇作为保护剂,冻干产品外观良好,表面平整、饱满,呈白色饼状,但复溶后粒径增大2倍,粒度分布显著变宽,出现了粒子聚集;以蔗糖、海藻糖、麦芽糖作为保护剂时,冻干前后粒径变化均较小,且复溶后透光性均较好,但三者之中加入海藻糖的制剂外观最佳,复溶时间最短且粒度分布最窄,因此以100 g·L-1的海藻糖作为冻干保护剂。

Table 5 Effect of cryoprotectants on BU-LP-NPs in the freeze-drying.表5 冻干保护剂种类对蟾毒配基脂质-蛋白杂化纳米粒冻干过程的影响
2.6 BU-LP-NPs的制剂学性质评价
2.6.1 形态观察
采用透射电镜(transmission electron microscopy,TEM)观察BU-LP-NPs的粒子形态。具体操作如下:取BU-LP-NPs样品溶液适量,加蒸馏水稀释至适宜浓度,取适量,滴至覆有碳膜的铜网上,以滤纸吸去多余液体,待完全干燥后,用体积分数2%磷钨酸负染1 min,自然干燥后,在透射电镜下观察纳米粒的形态,结果如图5。由图5可见,BU-LP-NPs呈圆整球形或类球形,粒度分布较均匀,粒子间未出现聚集。

Fig. 5 Transmission electron microscope (TEM) image of BU-LP-NPs (magnification 50 000×)图5 蟾毒配基脂质-蛋白杂化纳米粒透射电镜图(放大倍数 50 000×)
2.6.2 粒径及ζ-电位的测定
采用NicompTMPSS 380粒度/电位测定仪测定BU-LP-NPs的粒径(图6)和ζ-电位(图7)。制剂的平均粒径为 (82.4 ± 28.5) nm,PI为0.120,ζ-电位为-19.44 mV。实验中发现,由透射电镜观察到的粒径比动态光散射法(dynamic light scatter,DLS)测得的粒径略小一些,这是由于采用 DLS法测定时,粒子外围存在亲水层,该法测得的是粒子的水化径,而TEM制备样品时的干燥过程以及测定时的高真空条件可使粒径大幅度减小,从而导致两种方法测得粒径具有差异。
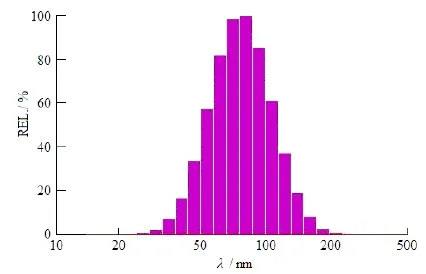
Fig. 6 The representative diagrams of Gaussian and Nicomp distribution of BU-LP-NPs图6 蟾毒配基脂质-蛋白杂化纳米粒粒径的高斯分布及尼康分布图

Fig. 7 The typical diagram of ζ-potential of BU-LP-NPs图7 蟾毒配基脂质-蛋白杂化纳米粒ζ-电位图
2.6.3 药物在制剂中的存在形式
药物在载体中的存在状态对制剂的稳定性具有重要影响,因此有必要确证药物在制剂中的存在形式。本研究采用差示扫描量热法(differential scanning calorimetry, DSC)和X-射线衍射法(X-ray diffraction, XRD)考察蟾毒配基在脂质-蛋白杂化纳米粒中的存在状态。
2.6.3.1 DSC分析
取适量样品粉末,置于铝盘中,以氧化铝作为参比物,在氮气流中以10 ℃·min-1的速率升温扫描,升温范围为30→300 ℃。分别测定蟾毒配基、空白脂质-蛋白杂化纳米粒、蟾毒配基脂质-蛋白杂化纳米粒(BU-LP-NPs)、蟾毒配基与空白纳米粒的物理混合物及冻干后的100 g·L-1海藻糖溶液样品的DSC图谱,结果见图8。由图8可见,蟾酥分离物在95.52、217.04和248.55 ℃分别存在一个平缓的吸热峰,可能由3种蟾毒配基分别产生,空白纳米粒在274.95 ℃存在一个吸热峰,物理混合物在95.56、248.83和269.40 ℃存在3个极平缓的吸热峰,分别可与蟾毒配基和空白纳米粒的吸热峰相对应,但其中蟾毒配基的吸热峰强度极弱,可能是由于混合物中蟾毒配基的比例过低或者测定过程中随着温度增加,药物与磷脂、白蛋白形成复合物所致。海藻糖在 102.63、127.17、201.35和259.22 ℃存在4个较为尖锐的吸热峰,而BU-LP-NPs仅在274.71 ℃处具有空白纳米粒的吸热峰,峰位和峰形基本没有变化,药物与海藻糖的吸热峰均消失。结果表明,蟾毒配基在脂质-蛋白杂化纳米粒中可能以无定形状态存在。

Fig. 8 DSC of bufadienolides (a), lyophilized blank nanoparticle (b), lyophilized BU-LP-NPs with 100 g·L-1trehalose (c), physical mixture of bufadienolides and blank nanoparticle (d) and 100 g·L-1trehalose solution after lyophilization (e)图8 蟾毒配基(a)、冻干后的空白纳米粒(b)、冻干后的蟾毒配基脂质-蛋白杂化纳米粒(含100 g·L-1海藻糖) (c)、蟾毒配基和空白纳米粒的物理混合物(d)及100 g·L-1海藻糖溶液冻干物(e)的DSC图谱
2.6.3.2 XRD分析
为了进一步考察药物在纳米粒中的存在形态,对冻干后的粉末样品进行X-射线粉末衍射分析。测定条件:CuKa石墨单色器,管压40 kV,管流30 mA,扫描速度4°·min-1,扫描步长0.03°,扫描范围(2θ)为0°~50°,测定结果见图9。由图9可见,蟾毒配基显示出较弱的衍射峰,空白纳米粒图谱呈现出典型的无定形衍射峰,物理混合物的图谱中同时存在蟾毒配基和空白纳米粒的衍射峰,强度均较弱,是二者衍射图谱的简单叠加。BU-LP-NPs衍射图谱中,蟾毒配基的衍射峰完全消失,而空白纳米粒的衍射峰基本没有变化,表明蟾毒配基在脂质-蛋白杂化纳米粒中以无定形状态存在,这与DSC的测定结果是一致的。

Fig. 9 XRD diffractogram of bufadienolides (a), lyophilized blank nanoparticle (b), lyophilized BU-LP-NPs with 100 g·L-1trehalose (c) and physical mixture of bufadienolides and blank nanoparticle (d)图9 蟾毒配基(a)、冻干后的空白纳米粒(b)、冻干后的蟾毒配基脂质-蛋白杂化纳米粒 ( 含100 g·L-1海藻糖 ) (c)、蟾毒配基和空白纳米粒的物理混合物(d)的X-射线衍射图谱
2.6.4 体外释放特性考察
纳米给药系统体外释放常用的研究方法有透析法、流通池法和反向动态透析法等[4]。考虑到透析法操作简便,且预试验已证明透析袋对蟾毒配基无吸附作用,因此采用动态膜透析法对BU-LP-NPs及蟾毒配基的500 g·L-1乙醇溶液(BU-S)进行体外释放度研究。蟾毒配基为水难溶性药物,为满足漏槽条件,需要在释放介质中加入有机溶剂,但加入过多有机溶剂与人体生理环境不符,因此在pH 值7.4的PBS缓冲液中加入2 g·L-1吐温-80作为释放介质。
操作方法:精密量取BU-LP-NPs和BU-S各2.0 mL,分别置透析袋(截留分子质量1.4×104u)中,两端用麻绳系紧,置于250 mL具塞锥形瓶中,加入100 mL释放介质。将锥形瓶置于(37±0.5) ℃的恒温水浴振荡器中,100 r·min-1振荡,分别于0.5、1、2、4、6、8、12、24、36、48和72 h取样100 μL,并补加同体积的释放介质,将取出的介质用HPLC法进样分析,计算BU-LP-NPs和BU-S在各个时间点的累计释药百分比(Q),结果见图10、11。

Fig. 10 The release profiles of B, C and R from BU-LP-NPs in pH 7.4 PBS containing 2 g·L-1tween-80图10 纳米粒中的3种蟾毒配基(B,C,R)在pH7.4值磷酸盐缓冲液(含2 g·L-1吐温-80)中的释放曲线
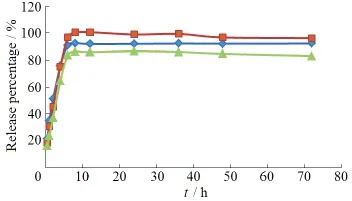
Fig. 11 The release profiles of B, C and R from BU-S in pH7.4 PBS containing 2 g·L-1tween-80图11 蟾毒配基溶液中的3种蟾毒配基(B,C,R)在pH值7.4磷酸盐缓冲液(含2 g·L-1吐温-80)中的释放曲线
由图10、11可见,BU-LP-NPs和BU-S在0.5 h内药物的释放量均小于40%,未出现突释现象,且2种制剂中蟾毒配基的释放速度均为C>B>R。制剂中的3种蟾毒配基在24 h时释放均已达到平台期,基本释放完全,而BU-S中的3种蟾毒配基在8 h时已基本释放完全,释放量分别为92.46%、100%和86.36%,表明BU-LP-NPs可延缓药物的体外释放。
将BU-LP-NPs的释放曲线分别用一级、Higuchi、Weibull和Ritger-Peppas方程拟合,其中Weibull方程的拟合度最好,蟾毒配基B、C和R的拟合方程及相关系数见表6。结果表明,BU-LP-NPs中药物的释放符合Weibull释放模型,其释放特征为初始阶段的快速释放及后期的缓慢释放,释放机制以扩散为主。

Table 6 The regression equation of B, C and R release rate from BU-LP-NPs in vitro表6 纳米粒中的三种蟾毒配基(B,C,R)体外释放曲线的回归方程
3 讨论
3.1 包封率测定方法的选择
目前测定纳米粒包封率的方法有多种,如葡聚糖凝胶柱色谱法、透析法、超速离心法、离心超滤法等,其原理都是利用一定的方法使游离药物与被包封药物分离,测定游离或包封药物的浓度,计算包封率。但凝胶柱色谱法费时费力,可导致纳米粒的稀释,不易观察混浊现象,且使药物浓度过低而难以检测;透析法操作时间长且稀释倍数大,透析过程中可能出现被包封药物泄漏的现象;超速离心法由于其离心力巨大,纳米粒易被破坏,使被包封的药物进入上清液,且小粒径纳米粒难以完全沉淀,使上清液中药物浓度偏高,导致测定结果不准确。实验过程中曾采用超速离心法(5×104r·min-1, 2 h)测定纳米粒的包封率,但纳米粒难以完成沉淀,使测得的结果偏低。离心超滤法由于超滤管的合理设计,可实现游离药物与纳米粒的有效分离,且操作简便、快速、离心力较小,不会造成纳米粒破碎,因此适于蟾毒配基蛋白纳米粒包封率的测定。
3.2 粒径及粒度分布的控制
当溶液中的微粒小于1 μm时,粒子的饱和溶解度受粒径大小影响,即大粒子溶解度小,而小粒子溶解度大,导致小粒子逐渐溶解同时大粒子逐渐变大,该现象称为奥斯特熟化(Ostwald ripening)。为了避免该现象,体系的粒度分布应较窄。作者所制备的BU-LP-NPs粒径及粒度分布均较小,可较好的避免奥斯特熟化现象,这可能是由于磷脂与白蛋白均可显著降低粒子的表面张力,在粒子表面形成牢固的乳化膜,较少药物的渗漏,从而抑制奥斯特熟化现象,且白蛋白在油水界面处构象伸展,具有一定的空间稳定作用,抑制了粒子间的聚集[9],从而保证BU-LP-NPs具有较好的稳定性。
3.3 冻干保护剂的选择
实验中考察的系列冻干保护剂中,海藻糖的冻干保护效果最好,可能与其结构与理化性质有关。海藻糖为非还原性二糖,可在冷冻干燥及低渗条件下保护生物膜及蛋白质的结构与功能,对蛋白质的空间结构起到稳定作用[10]。文献报道,海藻糖为环状结构,可与纳米粒的极性基团结合,形成致密的保护层,与链状分子相比,其空间抵抗力更大,因此可有效抑制粒子的聚集[11-12]。另外,海藻糖水溶液在冻干过程中不会析出晶体,而是形成黏度较大的玻璃态后固化,在纳米粒间提供无定形基质,其无定形态可维持囊泡的流动状态并降低囊泡表面张力,且可防止磷脂膜融合以及冰晶的形成对磷脂膜的机械破坏[13-14];同时海藻糖可与磷脂的极性基团形成氢键,代替水与磷脂间的氢键,使纳米粒嵌入海藻糖基质内部,从而保持纳米粒的形态与粒径[15]。
3.4 药物存在的形式
由DSC及XRD图谱推断,蟾毒配基在纳米粒中以无定形态被磷脂及白蛋白包裹。这可能是由于在旋蒸除去乙醇的阶段,溶解在乙醇中的蟾毒配基随着乙醇的蒸发不断析出,而磷脂的包绕作用及空间稳定作用抑制了蟾毒配基药物结晶的形成,从而使蟾毒配基在冻干制剂中以无定形态存在[16],并且在长期储存过程中可抑制蟾毒配基由无定形向结晶态转化,有利于提高制剂在储存过程中的稳定性。
4 结论
作者采用搅拌超声法制备了BU-LP-NPs,并单因素考察了处方及制备工艺,以最优处方及工艺制备的纳米粒外观圆整、粒径较小且分布均匀、产率及包封率均较高。确定了冻干工艺并以100 g·L-1海藻糖作为冻干保护剂,冻干产品复溶迅速,透光性好,且冻干前后粒径无明显变化。DSC及 X-射线结果显示,蟾毒配基在制剂中以无定形态存在,体外释放实验表明,BU-LP-NPs无突释效应,且可显著延缓药物的释放,制剂中3种蟾毒配基的释放均符合Weibull模型,释放机制以扩散为主。
[1] KRENN L, KOPP B. Bufadienolides from animal and plant sources[J]. Phytochemistry, 1998, 48(1): 1-29.
[2] STEYN P S, VAN HEERDEN F R. Bufadienolides of plant and animal origin[J]. Nat Prod Rep, 1998, 15(4): 397-413.
[3] LACKO A G, NAIR M, PROKAI L, et al. Prospects and challenges of the development of lipoprotein-based formulations for anti-cancer drugs[J]. Expert Opin Drug Deliv, 2007, 4(6): 665-675.
[4] KADER A, PATER A. Loading anticancer drugs into HDL as well as LDL has little affect on properties of complexes and enhances cytotoxicity to human carcinoma cells[J]. Journal of Controlled Release, 2002, 80(1-3): 29-44.
[5] RENSEN P C N, DE VRUEH R L A, KUIPER J, et al. Recombinant lipoproteins: lipoprotein-like lipid particles for drug targeting[J]. Advanced Drug Delivery Reviews, 2001, 47(2-3): 251-276.
[6] 苏德森, 王思玲. 物理药剂学[M]. 北京: 化学工业出版社, 2004:372-373.
[7] GAO H, CAO S, CHEN C, et al. Incorporation of lapatinib into lipoprotein-like nanoparticles with enhanced water solubility and anti-tumor effect in breast cancer[J]. Nanomedicine, 2013, 8(9): 1429-1442.
[8] GRIT M, UNDERBERG W J, CROMMELIN D J. Hydrolysis of saturated soybean phosphatidylcholine in aqueous liposome dispersions[J]. J Pharm Sci, 1993, 82(4): 362-366.
[9] 张辉, 霍美蓉, 周建平, 等. 伊曲康唑白蛋白纳米混悬剂的制备和表征[J]. 中国药科大学学报, 2008, 39(6): 510-514.
[10] MILLER D P, ANDERSON R E, DE PABLO J J. Stabilization of lactate dehydrogenase following freeze thawing and vacuum-drying in the presence of trehalose and borate[J]. Pharm Res, 1998, 15(8): 1215-1221.
[11] ZHANG L, LIU L, QIAN Y, et al. The effects of cryoprotectants on the freeze-drying of ibuprofen-loaded solid lipid microparticles (SLM)[J]. Eur J Pharm Biopharm, 2008, 69(2): 750-759.
[12] 李芳. 蟾毒配基静脉注射载药系统的研究[D]. 沈阳:沈阳药科大学, 2010: 25-30.
[13] MIYAJIMA K. Role of saccharides for the freeze-thawing and freeze drying of liposome[J]. Advanced Drug Delivery Reviews, 1997, 24(2/3): 151-159.
[14] CHEN C, HAN D, CAI C, et al. An overview of liposome lyophilization and its future potential[J]. Journal of Controlled Release, 2010, 142(3): 299-311.
[15] WANG D Q, HEY J M, NAIL S L. Effect of collapse on the stability of freeze-dried recombinant factor VIII and alpha-amylase [J]. J Pharm Sci, 2004, 93(5): 1253-1263.
[16] SIGFRIDSSON K, FORSSEN S, HOLLANDER P, et al. A formulation comparison, using a solution and different nanosuspensions of a poorly soluble compound[J]. Eur J Pharm Biopharm, 2007, 67(2): 540-547.
Preparation and in vitro evaluation of bufadienolides-loaded lipid protein hybridization nanoparticles
LI Wanqiu1, LIN Xia2, XU Hang3, TANG Xing3*
(1. College of traditional Chinese Materia medica, Shenyang Pharmaceutical University, Shenyang 110016, China; 2. School of Pharmacy, Shanghai Jiao Tong University, Shanghai 200240, China; 3. School of Pharmacy, Shenyang Pharmaceutical University, Shenyang 110016, China)
ObjectiveThe purpose of this study was to prepare bufadienolides-loaded lipid protein hybridization nanoparticles (BU-LP-NPs) by mixing ultrasonic method and to investigate whether the nanoparticles could delay the release of bufadienolides.MethodsSingle-factor experiments were applied to optimize the formulation and preparation process. Moreover, transmission electron microscopy was used to observe the morphology of nanoparticle and dynamic light scattering (DLS) method was employed to determine particle size and ζ-potential. Besides, the dispersion state of drugs in the formulation was characterized by differential scanning calorimetry (DSC) and X-ray diffraction method. The release behavior of drugs in BU-LP-NPs and bufadienolides solution (BU-S) was evaluated by dialysis method.ResultsBU-LP-NPs was round, and uniform with mean particle size of (82.4 ± 28.5) nm and ζ-potential of -19.44 mV. Drugs in formulation existed in amorphous form. Compared with BU-S, BU-LP-NPs had a significant performance of delaying the release of bufadienolides. The in vitro release of bufadienolides in BU-LP-NPs followed Weibull model and the releasing mechanism was primarily diffusion.ConclusionsThe lipid protein hybridization nanoparticles, prepared by mixing ultrasonic method, with endogenous HSA and egg lecithin as primary carriers and stabilizing agents, can significantly delay the in vitro release of bufadienolides.
Pharmaceutics; lipid protein hybridization nanoparticles; mixing ultrasonic method; bufadienolides; formulation optimization; preparation process; in vitro release
(本篇责任编辑:赵桂芝)
R94
:A
(2016)05-0160-15
10.14146/j.cnki.cjp.2016.05.003
2015-05-22
李晚秋(1988-), 女(汉族), 河北秦皇岛人, 硕士研究生,E-maillwq96@126.com;*
唐星(1964- ), 男(汉族), 陕西商县人, 教授, 博士, 博士生导师, 主要从事口服微粒缓控释制剂和中药现代化研究,Tel.024-23986343,E-mailtangpharm@sina.com。
