云南黑山羊全基因组重测序
2016-12-28邵庆勇洪琼花
兰 蓉,朱 兰,邵庆勇,洪琼花
(云南省畜牧兽医科学院,云南昆明650224)
云南黑山羊全基因组重测序
兰 蓉,朱 兰,邵庆勇,洪琼花*
(云南省畜牧兽医科学院,云南昆明650224)
采用Illumina Hiseq2000测序技术对由云南黑山羊具有代表性的个体构建的DNA池进行20X全基因组重测序,以期对云南黑山羊分子特征做出评价,并为云南黑山羊功能基因定位提供分子基础数据。结果表明:云南黑山羊可以检测到7 615 774个SNP、877 232个INDEL和40 005个SV。通过比对山羊参考基因组,并进行生物信息学分析,结果显示云南黑山羊位于外显子区域的SNP有35 902个,其中异义突变17 160个,同义突变18 920个;外显子区域的小INDEL有1 330个;位于内含子区域的SNP 1 695 420个,小INDEL 208 999,位于UTR3区域的SNP 16 106个,小INDEL 580个。研究结果基本阐明了云南黑山羊的分子特征,为后续功能基因的研究提供了强大的数据支撑,并为功能基因的定位提供新的思路和线索。
云南黑山羊;全基因组重测序;单核苷酸多态性;小片段插入缺失变异;结构变异
10.16863 /j.cnki.1003-6377.2016.05.003
近年来,随着基因测序技术的发展和进步,测序时间的缩短和成本的降低,科学家们使用全基因组测序技术获得大量物种的基因组序列。以此为基础,通过全基因组重测序技术(whole-genome resequencing,WGR)对已知基因组序列的不同个体进行基因组重测序和序列对比分析,可得到物种个体差异的遗传信息,包括大量的单核苷酸多态性位点(SNP)、拷贝数变异(CNV)、插入缺失位点(InDel)、结构变异位点(SV),最终获得生物群体的遗传特征。利用全基因组重测序技术,有助于快速发现与动植物重要性状相关联的遗传信息,开发相关全基因组遗传标记,加快分子育种,对科学家研究物种的遗传进化、自然选择、疾病研究具有重要的意义。
云南黑山羊是由云岭黑山羊与努比羊杂交培育而成的一个肉羊新品种,具有肉质鲜美、生长快、产羔率高的特点,它克服了云岭黑山羊生长慢、产羔率低的缺点,但保留了地方黑山羊独特的风味。本研究旨在通过对这一个培育新品种全基因组重测序,全面、快速、准确掌握其分子遗传特征,为这一品种的不断选育提高及开发利用奠定坚实的基础。
1 材料与方法
1.1材料
1.1.1 实验羊只
随机选取石林生龙公司种羊场云南黑山羊核心群种母羊3只。
1.1.2 血样
用真空采血管采集实验羊只颈静脉血2 m L,EDTA抗凝,-20℃保存备用。
1.1.3 试剂
DNA提取试剂盒、10×TE均购自大连宝生物公司,文库制备试剂盒(TruSeq DNA PCR-Free Library Preparation Kit)、目标富集试剂盒(HiSeq Rapid SR Cluster Kit)购自美国illumina公司,其它试剂均为国产分析纯。
1.2方法
1.2.1 DNA提取
按试剂盒blood genome DNA Extraction kit操作流程提取血样DNA,1×TE缓冲液溶解,保存于-20℃备用。
1.2.2 全基因组重测序
将3个样品基因组DNA等比例混合构成DNA池,随机打断,纯化所需长度DNA片段,在末端加上接头后,进行cluster生成,送至美吉生物用HiSeq 2000自动完成,实验流程如图1。
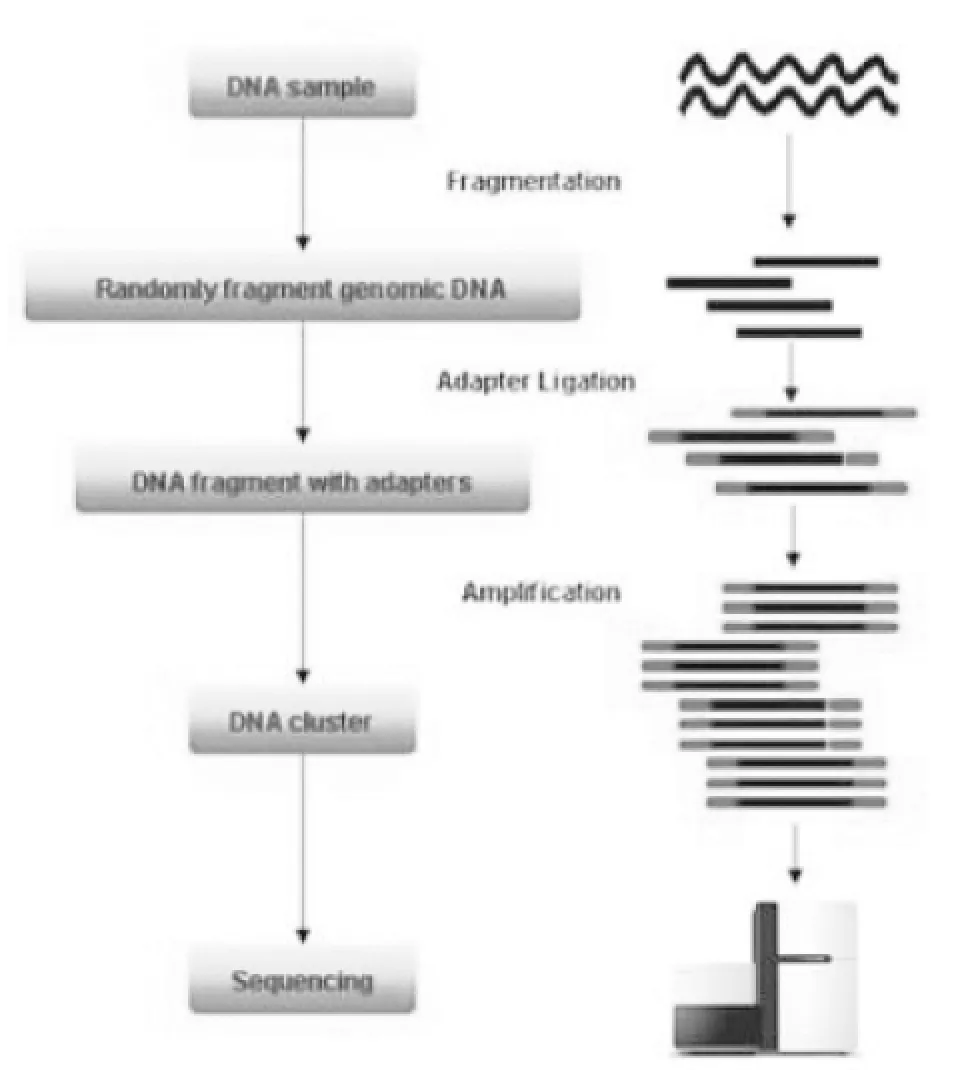
图1 重测序分析流程
1.2.3 原始测序数据质量剪切
云南省黑山羊的DNA通过Illumina测序技术进行paired-end(PE)测序后,可构建文库。但为了后续分析的准确性,需要对于Illumina原始测序数据要进行质量剪切,即剪切掉reads中的adapter序列、去除5’端含有非A、G、C、T的碱基,并对测序质量值<20的reads末端进行修剪。另外还需去除质量较低数据,包括去除含N比例高于10%的reads,以及去除adapter以及质量修剪后长度仍小于25bp的小片段。1.2.4生物信息分析:运用BWA[4]软件对测序得到reads与参考基因组序列进行比对,然后利用Picard-tools[5]去除PCR-duplication产生的测序reads,根据比对结果,计算出相对于参考基因组的测序深度和覆盖度。利用GATK[6]软件包进行SNP、small indel信息检测。对于检测到的突变位点,用samtools[7]提供的vcfutils工具以及GATK过滤掉测序深度和比对质量值较低的位点。用BreakDancer[8]软件对样品的SV进行检测。利用annovar[9]软件和参考序列的gff信息进行注释,得到SNP、indel、SV的注释信息。生物信息分析流程见图2。
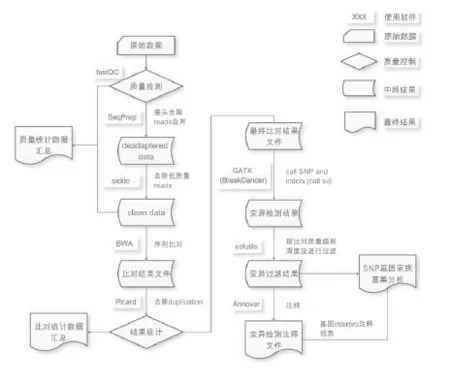
图2 生物信息分析流程
2 结果
本研究采用Illumina Hiseq2000测序技术完成了云南黑山羊全基因组重测序,构建了一个Illumina PE文库(300bp),对获得的测序数据进行质控后利用生物信息学分析手段完成样品的全基因组重测序分析。
2.1原始测序数据统计
测序得到原始图像数据经过Base Calling转化为序列数据,结果以FASTQ文件格式来存储。FASTQ文件为最原始的数据文件,文件包含测序reads的序列信息以及测序质量信息。格式如表1。
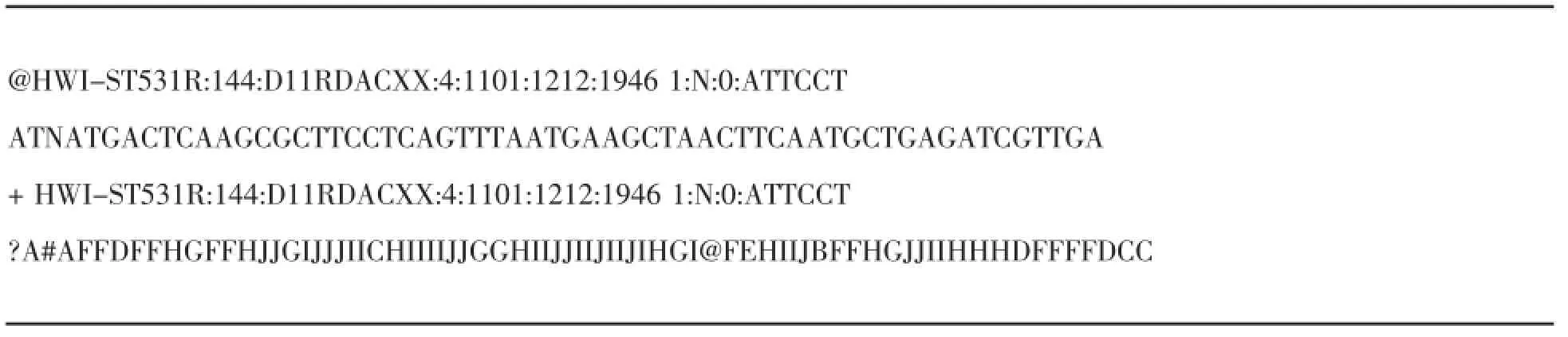
表1 F AS TQ原始数据格式示范
2.2原始测序数据质控
利用生物信息统计学方法,对所有测序reads的碱基分布和质量波动进行统计,从宏观上可以直观的反映出测序样本的测序质量和文库构建质量。原始数据碱基组成分布图(又称为GC偏差图)见图3,原始数据碱基质量分布图见图4。

注:横坐标是reads碱基坐标,纵坐标是read的碱基质量(Solexa Scale:40=Highest,-15=Lowest),图中垂直红线”Ⅰ”指定的范围是所有reads碱基的综合质量,红色垂直方块是质量的四分位值范围,加粗水平黑线是质量值的中位数。图4原始数据碱基质量分布图
2.3测序数据统计结果
质量剪切前后的数据的测序reads数、总碱基数、Q20%、文库平均插入长度、平均测序深度分别进行统计,结果见表2。
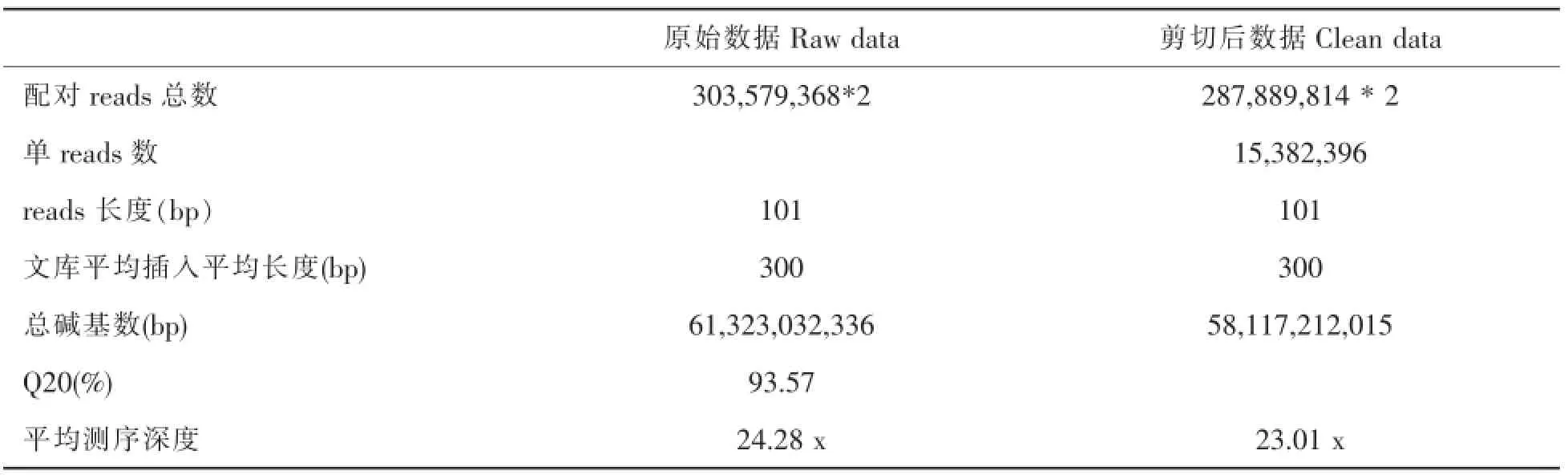
表2 测序数据统计表
2.4突变预测及注释结果统计
2.4.1 比对结果数据统计
本研究中,我们以山羊的基因组序列[10]作为参考,利用BWA[4]比对软件将质控后的测序片段比对回参考基因组[10,11],利用Picard-tools[5]去除PCR-duplication产生的测序片段,比对结果见表3。

表3 比对结果数据统计表
2.4.2 SNP、sm all indel检测和注释
根据最终的比对结果,利用GATK软件获得云南黑山羊群体SNP信息和群体InDel信息,见表4。采用Annovar[9]程序结合本项目山羊的gff基因注释信息对得到的SNP和small indel进行注释,见表5。表3-5为SNP、small indel注释结果统计表,图为SNP、small indel注释结果统计图。

表4 群体单核苷酸多态性、插入缺失位点数据统计表

表5 单核苷酸多态性、插入缺失位点注释结果统计表
2.4.3 外显子区域SNP和small indel突变位点对蛋白翻译影响
对于在exonic区域的SNP和small indel位点,变位点对蛋白翻译所产生的影响,统计结果见表6。

表6 外显子区域突变位点对蛋白翻译影响结果统计表
2.4.4 染色体结构变异(SV)分析
结构变异(SV)通是指长度在几Kb至几Mb染色体变异,类型包括:缺失(DEL)、插入(INS)、倒位(INV)、染色体内易位(ITX)和染色体间易位(CTX)。用软件BreakDancer(v1.1.2)[8]检测群体的SV,统计结果见表7。

表7 结构变异预测结果统计表
3 讨论
变异一直是遗传学研究的对象,而各种类型的变异基础是基因组变异。二代测序技术的出现,使得基因检测变得更加快速,准确和规模化,极大的加快了人类对基因组变异的研究。本研究即是在这样的技术背景下开展,因而获得了分布于云南黑山羊全基因组的大量变异信息,为这一品种的后续研究提供了重要的基因组变异基础信息。
本研究所检测的SNP、INDEL和SV显示基因组不同程度的遗传变异。云南黑山羊的SNP达到了7,615,774个,INDEL 877,232,SV 40005,分别占山羊的参考基因组[11](大小为2,661,316,841 bp)的2.86%、0.33%和0.015%,这足以说明点突变是基因组中最普遍存在的变异类型。然而也正由于SNP巨大的信息量,假如参考基因组注释信息不完全公开,要进行快速的功能基因定位难度较大,与之相比,具有投入成本以及检测准确性等方面优势的低频变异,如INDEL及SV等,在遗传学、生态学、癌症学、发育学等领域有着广阔的应用前景。
田大成等2008年提出了“INDEL诱导自发突变机制假说”[12],认为基因组各区域的突变率各不相同,INDEL的数量和密度决定了自发突变的数量,INDEL的诱导可能是生物多样性的最初变异来源,对INDEL的选择在很大程度上实现了自然选择,从而在很大程度上决定了自发突变率的高低。这一观点在很大程度上说明了INDEL在遗传变异研究中的重要性,也提示在本研究中所检测到的大量INDEL存在的合理性,为今后的研究重点指出了方向。另外,本研究注释了位于外显子区域的1 444个INDEL,其中移码突变占68.4%,占比较高。移码突变是DNA链中缺失一个或几个碱基,或者是插入一个或几个碱基,从而使突变位点以后的碱基序列发生变更,致使三联密码转录和翻译时发生较多的遗传变异,本研究中注释到大比例的INDEL移码突变,暗示我们云南黑山羊具有丰富的遗传变异分子基础,有必要重点、深入研究云南黑山基因组中存在的INDEL移码突变,这对云南黑山羊新基因的发掘与利用具有重要意义。
SV通常是指长度在几Kb至几Mb染色体变异,是基因组变异的主要来源,在某些区域上,甚至SV的形成速率要大于SNP的速率,并与疾病临床表型密切关联。本研究中注释到的SV中,最多的是缺失结构变异,占比为79.9%,而在已有的人类疾病研究表明,染色体缺失变异是最为常见的变异类型,它通常是伴随染色体插入变异而大量产生[13],可能会使基因组的稳定性受到破坏,影响基因间相互作用的关系,也可能会改变基因排列位置关系,从而导致基因所决定或控制的功能丧失或改变。这些研究结果说明染色体缺失结构变异对生物体遗传效应的改变作用极大。本研究中检测到的大量缺失结构变异又是与山羊的何种性状、何种疾病相关呢?这为我们今后的研究工作提出了新的课题。
4 结论
本研究阐明了云南黑山羊的分子特征,为后续功能基因的研究提供了强大的数据支撑,并为功能基因的发掘和定位提供了新的思路和线索。
[1]Rabbani B,Tekin M,Mahdieh N.The promise of whole-exome sequencing in medical genetics[J].Hum Genet,2014,59(1):5-15.
[2]Boycott KM,Vanstone MR,Bulman DE,MacKenzie AE.Rare-disease genetics in the era of nextgeneration sequencing:discovery to translation[J].Nat Rev Genet,2013,14(10):681-691.
[3]O'Roak BJ,Deriziotis P,Lee C,Vives L,et al.Exome sequencing in sporadic autism spectrum disorders identifies severe de novomutations[J].NatGenet,2011,43(6):585-589.
[4]LiH,Durbin R.Fastand accurate long-read alignmentwith Burrows-Wheeler transform[J].Bioinformatics, 2010,26(5):589-595.
[5]http://broadinstitute.github.io/picard/
[6]http://www.broadinstitute.org/gatk/
[7]LiH,Handsaker B,Wysoker A,etal.The Sequence Alignment/Map formatand SAMtools[J].Bioinformatics, 2009,25(16):2078-2079.
[8]Fan X,Abbott TE,Larson D,et al.BreakDancer:Identification of Genomic Structural Variation from Paired-End Read Mapping[EB/OL].Curr Protoc Bioinformatics,2014,doi:10.1002/0471250953.bi1506s45.
[9]Wang K,LiMY,Hakonarson H.ANNOVAR:functional annotation of genetic variants from high-throughput sequencing data[J].Nucleic Acids Res,2010,38(16):e164.
[10]http://goat.kiz.ac.cn/GGD/download9.htm
[11]Dong Y,Xie M,Jiang Y,et al.Sequencing and automated whole-genome opticalmapping of the genome of a domestic goat(Capra hircus)[J].Nat Biotechnol,2013,31(2):135-41.
[12]Tian DC,Wang Q,Zhang PF,etal.Single-nucleotidemutation rate increases close to insertions/deletions in eukaryotes[J].Nature,2008,(455):105-108.
[13]Urban M,King R,Hassani-Pak K,et al.Whole-genome analysis of Fusarium graminearum insertional mutants identifies virulence associated genes and unmasks untagged chromosomal deletions[J].BMC Genomics,2015,16(1):261.
Whole-genome Resequencing in Yunnan Black Goat
LAN Rong,ZHU Lan,SHAOQing-yong1,HONGQiong-hua*
(Yunnan Institute of Animal Science and Veterinary,Kunming 650224 China)
To evaluatemolecular characteristic of Yunnan black goatand to provide basicmolecular data for its functional gene assignment,20X whole-genome resequencing was done by Illumina Hiseq2000 in the DNA poolof representative individuals.Itwas found that7 615 774 single nucleotide polymorphism(SNPs),877 232 insertion-deletion(InDels)and 40 005 structure variation(SVs)were detected in the genome of Yunnan black goat.Compared with the reference genome,and by analysis with bioinformatics,SNPs located in exon area were 35 902,ofwhich 17 160 with missense mutation and 18 920 with synonymousmutation;small INDELs in such area is 1 330;SNPs in introns area were 1 695 420,small INDELswere 208 999;and SNPs in UTR3 were 16 106,and small INDELs were 580.This study indicates the molecular characteristic of Yunnan black goat and provides essential information for further research on functional genes and new thought and clue for mapping functional genes.
yunnan black goat;whole-genome sequencing;SNP;InDel;SV
S813.8
:A
:1003-6377(2016)05-0011-07
云南省科技计划项目(2014BB014);国家现代农业产业技术体系建设(CARS-39)
兰蓉(1969-),女,云南人,副研究员,硕士,从事家畜分子遗传育种研究。Email:rtlankitty@163.com
洪琼花,研究员,Email:yxh7168@126.com
2016-04-20,
:2016-04-25
