固定漆酶聚苯胺-CoC2O4纳米复合物修饰电极的直接电化学
2016-12-20杨阳霍文珊周政张琪曾涵
杨阳 霍文珊 周政 张琪 曾涵
(新疆师范大学化学化工学院,新能源材料化学实验室,乌鲁木齐830054)
固定漆酶聚苯胺-CoC2O4纳米复合物修饰电极的直接电化学
杨阳霍文珊周政张琪曾涵*
(新疆师范大学化学化工学院,新能源材料化学实验室,乌鲁木齐830054)
采用循环伏安法、微分脉冲伏安法、交流阻抗谱以及计时电流法等电化学方法,结合红外光谱、紫外-可见分光光度法、原子力显微镜、透射电子显微镜以及原子吸收光谱等辅助手段,表征了固定漆酶的聚苯胺-草酸钴纳米复合物的化学组成、结构和形貌,测试了纳米复合物固酶前后的导电性能的变化,研究了纳米复合物修饰电极上固定漆酶的直接电化学行为,评估了该电极的催化氧还原效能以及作为电化学传感器检测氧分子的性能。实验结果表明该电极在不含电子介体的溶液中以酶活性中心T2作为首要电子受体,将得到电子传递给化学吸附的氧气使其被电还原,其表观电子迁移速率为0.017 s-1,且具有良好的催化氧还原性能(氧还原起始电位:460 mV vs NHE,转化氧分子为水的表观速率常数为2.6×10-4s-1),酶电催化氧还原为水分子步骤为反应的速控步。该电极作为电化学传感器对氧具有极低检测限(0.20μmol·L-1),宽线性响应范围(0.4~7.5μmol·L-1)以及对底物高亲和力(KM=122.4μmol·L-1)等优势。
聚苯胺-草酸钴纳米复合物;漆酶;直接电化学;催化氧还原反应;电化学传感器
0 引言
酶燃料电池是一种新型,对环境友好的能量来源,一般认为限制酶燃料电池能量输出性能的最重要因素在于阴极氧还原反应,而提高氧还原反应效率的方法除了提升固酶载体对氧的亲和力,化学吸附氧的性能以及氧在介质中的传质动力学之外,更重要的是要改善酶-导电基体间的电子迁移性能[1]。漆酶(Laccase,简称为Lac)可以在催化酚类,有机杂环化合物氧化的同时,按照四电子机制将化学吸附的氧分子转化为水分子而不经过生成过氧化氢的中间过程[2]。Lac由于具有较高氧化还原式电位,对底物选择性高以及较快的催化反应速率等优势,从而成为当前研究最多的一种酶基燃料电池阴极催化剂[1-4],但Lac活性中心由于结构非常复杂且被不导电的多肽链包覆在蛋白质内部,难以实现酶-电极间直接电子导通[5]。迄今为止文献报道实现酶-电极间有效电子导通的方法仍然存在一些缺陷:使用电子中介体会降低电池的输出能量密度,小分子有机物作为电子介体对生物体有毒副作用且易变性[6-7],稳定性较差,不适宜作为体内电化学器件组分使用[7];制备氧化还原水凝胶的工序复杂,成本高,机械稳定性和pH值耐受性都不够好,加上制备过程中需要使用贵重金属,也不适合在生物体内使用;而基于含大共轭π键的芳香化合物修饰纳米器件吸附酶的酶基电极也存在不足:制备电极时使用有毒且性质不稳定,易燃烧或爆炸的化合物,电流输出密度不高,催化剂无法回收,长期使用稳定性欠佳等[8-11]。一般认为以直接的方式实现酶-电极间有效电子迁移是提高酶燃料电池和酶基电化学传感器性能的最有效途径[12-13],常见的方法包括将酶分子固定在表面含有能和酶活性中心发生某些有效相互作用的载体,或者以自组装的方式将某些既能在导电基体表面可逆得失电子,又可以与酶活性中心进行有效电子交换的官能团或分子与酶分子以静电吸引,化学偶联或配位等方式整合于导电基体。这些载体包括金属氧化物纳米管[3],碳纳米管[4],金属含氧酸盐层状纳米片[12],以及水凝胶-无机纳米材料复合物[13]等。目前文献[8-10,14-15]所见多数能够实现酶-电极间直接电子迁移的Lac基电极,在没有外加介体存在时一般观察不到表征酶活性中心发生电子迁移的电化学信号,这就给深入研究固酶电极催化反应机制带来一定的困难。定量研究固酶电极结构参数对固酶电极催化底物反应性能的影响,电极表面固定酶分子催化底物的反应动力学,反应决速步和限制固酶电极催化性能的主要因素的确定依然是制备高性能酶燃料电池亟待解决的难题。聚苯胺因其合成制备简易,结构易于调控,导电性能优越等优势,在二次电池电极材料以及传感器等领域有着广泛的应用[16],文献[16-17]研究表明聚苯胺或聚苯胺衍生物接枝多壁碳纳米管对氧还原反应有着良好的催化性能,但其缺陷在于长期使用性能较差。由于聚苯胺的导电性能还不足以和普通电子导体或离子导体相竞争,因此将导电聚合物与无机金属纳米粒子复合是一个合理的选择。这种复合物结合了两者的优点从而具有独特的力,光,电,磁,热以及化学活性[18]。由于聚苯胺含有共轭大π键,易于依靠疏水-疏水作用接近Lac分子活性位附近的疏水分子结合位,因此被吸附的酶分子有较高概率实现酶活性中心与导电基体之间的直接电子迁移[19]。草酸钴(CoC2O4)纳米棒具有良好的力学性能和导电性能,其包含的羧基可与Lac分子表面氨基发生化学偶联,聚苯胺分子中的亚氨基氮原子与酶活性中心Cu2+之间可能存在的配位作用加上前述的疏水-疏水作用,聚苯胺-草酸钴纳米复合物可以依靠这三种作用的协同效应将酶分子稳定地固定在载体表面。这种协同作用也可能利于实现酶-导电基体之间的直接电子迁移。尽管如此,直接将这类纳米复合物滴涂在基底电极表面并不能获得稳定的纳米复合物修饰电极,仍然需要黏度较高,延展性较好的高分子聚合物作为成膜剂将固酶纳米微粒牢固地附着在电极表面。目前最常用的高分子成膜剂是壳聚糖及其衍生物,这类化合物不导电,对生物分子具有良好的亲和力。尽管部分这类化合物可以吸附氧分子,但研究表明这类化合物单独使用并不能够有效催化氧分子的电还原[6]。
本文以含有刚性芳环结构的聚苯胺(PAn)与CoC2O4混合所得纳米复合物作为固定Lac载体,制备了固定Lac基电极,以电化学技术结合光谱学方法研究了此固酶电极的直接电化学行为及催化氧还原性能,评估了其作为氧电化学传感器的性能。这些研究不仅为研究高性能酶燃料电池提供了新的思路,也有利于进一步了解酶催化氧还原机制和活性中心在催化反应中发生的结构变化,为开发仿生生物电子器件提供有益的探索经验。
1 实验部分
1.1仪器和试剂
云芝漆酶(Lac,源自Trametes Versicolor,分子量68 000),胆红素氧化酶(BOD,源自M.Verrucaria.分子量79 000),2,6-二甲氧基苯酚(DMP)购自美国Sigma化学试剂有限公司,直接使用无需进一步纯化;壳聚糖(脱乙酰度不小于90%,分子量250 000,简写为CTS),购自上海升耀生物技术有限公司;二水合草酸钴(CoC2O4·2H2O)、草酸(H2C2O4·2H2O)和其他常规化学试剂均为分析纯,购自国药集团化学试剂有限公司。实验过程中使用的缓冲溶液为0.2 mmol·L-1的磷酸盐缓冲液(PBS),溶液pH值籍调节磷酸氢二钠和柠檬酸溶液体积比例来调控;所有溶液均用Milli-Q超纯水配制。N2,O2(优级纯,来自南京特气)。2K15型高速离心机(德国Sigma公司),BRUKER TENSOR-27型红外光谱仪(德国BRUKER公司,KBr压片),日立z2000型原子吸收光谱仪(日本HITACHI公司,主机:单光束火焰/石墨炉原子吸收分光光度计,光谱范围:190~900 nm),U-2810型紫外可见分光光度计(日本岛津公司,比色皿厚度1 cm),透射电镜照片以H-800型透射电子显微镜(日本日立JEOL公司,加速电压:200 kV)拍摄,CSPM-5500型原子力显微镜(中国广州本原纳米仪器有限公司),采用“轻敲”模式(tapping mode);Zahner Zennium电化学工作站(Kronach,德国),CHI-1140A型电化学分析仪(CHI Inc,上海辰华),玻碳电极(GCE,直径3 mm,购自天津艾达恒晟工贸有限公司),饱和甘汞电极作为参比电极,对电极铂丝电极自制。工作电极使用前先以3500#砂纸,1.0和0.5μm氧化铝粉浆抛光,再用丙酮和三次重蒸水超声清洗各2次,每次2 min。文中如无特殊说明,所有的电极电位均为相对于标准氢参比电极(NHE)而言。
1.2聚苯胺-CoC2O4纳米复合物的制备
纳米草酸钴(CoC2O4)的制备过程参考文献[18]简述如下:将1.0 mmol CoC2O4·2H2O置于玛瑙研钵中磨细,加入1.0 mmol研细的草酸,研磨反应2 h后转移到圆底烧瓶中,加入50 mL水于70℃水浴中加热反应3 h,产物用超纯水洗涤到中性,干燥。
将制备的纳米CoC2O40.089 g超声分散在50 mL草酸(0.5 mol·L-1)溶液中。加入0.75 mL苯胺单体,超声40 min,按与单体1:1的物质的量之比加入氧化剂过硫酸铵,继续超声50 min,产物过滤,用超纯水和无水乙醇洗涤至滤液无色,55℃下真空干燥24 h,得到目标产物PAn-CoC2O4纳米复合物。
1.3固酶纳米复合物修饰电极的制备,直接电化学及催化氧还原性能
称取2 mg PAn-CoC2O4纳米复合物加入1 mL pH=4.4的磷酸盐缓冲液,超声分散30 min形成均匀稳定的分散相,向其中加Lac 1 mg,磁力搅拌30 min后放置于0℃冰箱中冷藏12 h。将固酶复合物于8 000 r·min-1下离心沉降20 min,倒去上清液后以少量PBS清洗沉积在离心管底部的固酶复合物3次,于同样转速下离心沉降,移除上清液后保留离心管底部的固酶复合物,收集上清液和滤液,将两者合并后,会同溶解Lac的母液一并送去测定铜离子浓度,便可参照文献[20]报道的石墨炉原子吸收法测定固酶复合物中固载Lac的量(mg·g-1,单位质量载体担载酶的质量),按前文[21]给出公式计算Lac固定百分率和载体对Lac的担载量。取5μL固酶纳米复合物滴涂到预处理过的GCE上,再抽取2μL CTS(质量浓度为1.5%)的冰乙酸溶液(冰乙酸质量浓度为3.0%)滴涂到玻碳电极表面,扣上烧杯于4℃冰箱内干燥6 h便可得到固定Lac基玻碳电极,缩写为Lac/PAn-CoC2O4/GC,类似地可以制备对比电极PAn-CoC2O4/GC。
采用紫外-可见分光光度法(UV-Vis)测定PAn-CoC2O4纳米复合物固定Lac前后的吸收光谱变化的具体方法如下:按照本节所述方法分别制备固定/未固定Lac的PAn-CoC2O4纳米粒子复合物,随后分别移取这些纳米粒子复合物200μL并均匀地涂覆在铟锡氧化物(ITO)玻璃片上,待干燥后插入比色槽中进行测定。所有测定的波长均在190~800 nm范围内,室温下进行。复合载体固定漆酶的比活力测定参照前文[21]方法,称取制备的固酶复合物5 mg与3mL pH值为4.4的PBS和100μL的10 mmol·L-1DMP混合液共混1 min后,测定室温下溶液中DMP氧化产物2,6-二甲氧基苯醌在470 nm处的吸光度,记录吸光度-时间关系曲线。2,6-二甲氧基苯醌的摩尔吸光系数为49 600 L·mol-1·cm-1,定义酶活力单位为1 min内氧化1μmol DMP所需要漆酶的量(1U)。电极的活性表面积按照文献[22]给出的方法,以K3[Fe (CN)6]作为探针进行标定,根据不同扫描速率下获得电流-电压曲线中阴阳极峰电流平均值与扫速平方根关系曲线拟合所得的斜率,便可测得PAn-CoC2O4/ GC的活性表面积为0.10 cm2。所有电化学测试在常规三电极电解池中进行。以Lac/PAn-CoC2O4/GC作为工作电极,不含任何电子介体的PBS缓冲液(pH= 4.4)作为电解质溶液。电化学阻抗谱(EIS)测试是在开路电压~235 mV,频率范围0.1~105Hz,交流激励信号幅值为5 mV的条件下进行;微分脉冲伏安法(DPV)曲线的测定是在电位范围-0.4~0.6 V,阶跃电位5 mV的条件下进行;研究固酶电极直接电化学行为实验中,实验前先向溶液中鼓泡通入N2除氧至少30 min,循环伏安法(CV)测试过程中电解液上方通入N2使溶液保持惰性气氛(所有电流-电压i-E曲线均为稳态下记录,即以扫描第10圈为准)。在催化氧还原实验中,实验前先向PBS缓冲液中不断鼓泡通入高纯O2至少15 min使溶液为O2饱和,实验中溶液上方维持O2气氛,所有测定均是在(25.0±0.4)℃下进行。文中给出的电流密度为测定的催化电流对电极的活性表面积归一化所得。以出现极限扩散电流时的电位处的电流差|iOxygen-iNitrogen|来表征固酶电极催化氧还原活力
1.4固酶纳米复合物修饰电极作为氧电化学传感器的性能
采用时间-电流曲线法(t-C)评估Lac/PAn-CoC2O4/GC对氧气的传感性能。t-C曲线系固酶电极在含有一系列不同O2浓度的PBS溶液中(pH=4.4)于恒定电压下记录响应电流-时间关系曲线而得到,这一系列不同O2浓度的PBS溶液系向N2气饱和的PBS溶液中加入不同体积的空气饱和PBS溶液(氧气浓度大约是260μmol·L-1)而制得,氧还原电位参考文献[6],以极限扩散电流对应电位作为工作电位。
2 结果与讨论
2.1PAn-CoC2O4纳米粒子复合物的形貌和结构表征
图1为CoC2O4·2H2O,PAn和PAn-CoC2O4纳米复合物的傅里叶变换红外光谱图(FTIR)。从图1可以看出:PAn-CoC2O4纳米复合物的FTIR中不但含有CoC2O4·2H2O的特征吸收峰,也含有PAn的特征吸收峰,除了Co-O的特征吸收峰由于复合物中共轭苯环存在而发生蓝移(分别位于591.5和674.1 cm-1)之外,还可以观察到一个新的吸收峰,位置在543.1 cm-1附近,这个吸收峰对应于-NH-的面外弯曲受到Co-N之间的作用影响,使其发生了蓝移,这间接证明纳米CoC2O4与聚苯胺之间发生了某种程度的化学作用。

图1 CoC2O4·2H2O,PAn和PAn-CoC2O4纳米复合物的FTIR谱图Fig.1 FTIR spectra of CoC2O4·2H2O,PAn and PAn-CoC2O4nano-composite
图2为PAn-CoC2O4纳米复合物薄膜固酶前后的UV-Vis对比图。从图2可以看出:固酶纳米复合物薄膜在560 nm附近出现一个对应于纳米复合物诱陷Lac活性位T1氧化态Cu2+d-d配位跃迁吸收峰,但此吸收峰位置与游离Lac的Cu2+d-d配位跃迁吸收峰(605 nm)[23]相比负向移动了45 nm,表明酶分子中Cu2+的d-d配位跃迁吸收峰在Co2+-N(聚苯胺或Lac分子中组氨酸残基中的N原子)和/或Cu-N配位作用的影响下,配体场较强引起d轨道分裂能增大,导致吸收峰发生了负移;此外200和280 nm的2个尖锐吸收峰对应于复合物中PAn芳环的强吸收带。这一现象暗示固酶载体中导电聚合物结构单元中杂原子与固定Lac活性中心Cu离子之间的相互作用对游离酶T1活性中心氧化态Cu离子固有配位构型产生了明显影响。值得说明的是,这种固酶纳米复合物所固定的Lac比活力为1.05 U·mg-1,大约保留了游离Lac比活力(1.52 U·mg-1)的70%,相对于文献[24]报道的纳米多孔金固酶复合物的比活力(0.83 U·mg-1)具有更高的催化活力。这一结果表明虽然酶-载体间的相互作用对酶活性中心离子的配位构型产生了一定的影响,但并没有过多地影响酶活性中心的催化反应机制,即酶分子内电子迁移桥链并没有被破坏。

图2 PAn-CoC2O4纳米复合物固酶前后的UV-Vis谱图Fig.2 UV-Vis spectra of PAn-CoC2O4nano-composite with Laccase(solid line)and without enzyme (dashed line)
图3为纳米CoC2O4和PAn-CoC2O4纳米复合物的原子力显微镜(AFM)照片,提供了它们的表面形貌和三维立体图像细节。从图3可以看出,纳米CoC2O4具有不规则的几何形状和凹凸不平的表面,这种具有不平整表面的形貌是由于制备的CoC2O4纳米棒通过堆积、重叠和挤压形成的;而PAn-CoC2O4纳米复合物则呈现出较为光滑的表面形貌,并能看到明显的层次分布,因此可以推测PAn在聚合过程中将具有不规则外形的CoC2O4纳米粒子包覆起来,因此具有均匀且平整的表面形貌。

图3“敲击”模式下的纳米CoC2O4(a)及PAn-CoC2O4纳米复合物(b)的AFM形貌图Fig.3 AFM topography images under tapping-mode of nano-CoC2O4(a)and PAn-CoC2O4nano-composite(b)
图4分别为纳米CoC2O4(A),PAn-CoC2O4纳米复合物(B)和固定Lac的PAn-CoC2O4纳米复合物(C)的透射电子显微镜(TEM)照片。从图4可看出:制备的纳米CoC2O4呈现出不规则的棒状结构,其中还有部分棒状纳米CoC2O4相互聚集,出现了更大、更宽的柱状结构,这可归因为相互接近的纳米CoC2O4依靠表面COOH间的氢键作用导致的团聚成簇(图4A);PAn-CoC2O4纳米复合物的形貌则呈现出整体上不规则的形态:部分保持纳米纤维形态,而另一部分纳米粒子相互缠绕形成无定形的团簇结构(图4B);而固定Lac的PAn-CoC2O4纳米复合物的形貌与前两者皆有明显差异:原有的棒状结构大部分消失了,由于纳米复合物表面被蛋白质分子包覆,因此可以观察到复合物周围存在柔性的膜状物质,在靠近纳米复合物表面可以发现存在颜色较深的刺状突起(图 4C),这暗示部分酶分子以某种特定构型被固定于纳米复合物表面,这可归因于聚苯胺中的芳环结构与Lac分子活性中心T1附近的疏水分子结合位之间的π-π堆积效应。
此外,测定结果表明该纳米复合物与传统的固酶载体比如介孔硅微球[25]以及多孔纳米金粒子[24]相比,其对Lac担载量(20.3 mg·g-1)和固酶百分率(35.0%)居中,这可能是因为PAn-CoC2O4纳米复合物对Lac的吸附具有特定的方向选择性(可归因于前述π-π堆积效应以及CoC2O4和Lac分子表面氨基酸残基之间的氢键作用的协同效应),与其他固酶纳米复合物显示表面较为光滑的无规则团簇的形貌有较大差异[26],故前者相对于后者只能固定较少量的酶分子,而且稳定性也不如后者。当固酶复合物以离心沉降形式分离上清液和固酶载体时,转速较高时一部分吸附不紧密的表面酶分子就会从固酶载体上脱落(在上清液中加入DMP,观察到由于泄漏的酶催化DMP氧化导致溶液颜色迅速变红这一现象)。

图4 纳米CoC2O4(A),PAn-CoC2O4纳米复合物(B)及固定Lac的PAn-CoC2O4纳米复合物(C)的TEM照片Fig.4 TEM images of nano-CoC2O4(A),PAn-CoC2O4nano-composite(B)and PAn-CoC2O4nano-composite with immobilized Lac molecules(C)

图5 裸GC电极(a),PAn-CoC2O4纳米复合物修饰电极(b)以及固定Lac的PAn-CoC2O4纳米复合物修饰电极在5 mmol·L-1K3[Fe(CN)6]+0.1 mol·L-1KCl混合液中以100 mV·s-1扫速扫描所得CV曲线,附图为上述电极在相同电解质溶液中的Nyquist曲线Fig.5 CV curves of bare GC electrode(a),basal electrode modified by PAn-CoC2O4nano-composite(b)and basal support over-coated by PAn-CoC2O4nano-composite with entrapped Lac molecules(c)in mixed solution of 5 mmol·L-1K3[Fe(CN)6]+0.1 mol·L-1KCl recorded at 100 mV·s-1,inset: Nyquist plots at all electrodes described previously in the same electrolyte
2.2固酶纳米复合物修饰电极的直接电化学及催化氧还原性能
2.2.1固酶纳米复合物修饰电极的直接电化学
图5及其附图分别是裸GC电极(a),纳米复合物修饰电极(b)以及固定Lac纳米复合物修饰电极在5 mmol·L-1K3[Fe(CN)6]+0.1 mol·L-1KCl混合液中扫描所得CV曲线,以及这3个电极在此电解液中所得EIS谱图。从图5及其附图可看出:纳米复合物修饰电极与裸GC电极的CV曲线相比,氧化还原峰电位差ΔEP有一定程度的降低(从121.5 mV下降到了115.3 mV),阴阳极峰电流比值ip,a/ip,c接近(分别是0.967和0.989),这表明纳米复合物增大了电极的活性表面积(活性面积增加了大约30%)并提高了电极上电活性物种反应的可逆性;而固定Lac的纳米复合物修饰电极的CV相对于电极固定酶分子之前,不但氧化还原峰电流和电极活性表面积有明显的下降(下降了大约48.8%),而且ΔEP明显增大(140.2 mV),这不仅表明电极反应的可逆性有所下降,而且电极表面活性面积也有显著的降低,这可归因于电极上负载的部分不导电的酶分子阻碍了电活性物种在导电基体表面的氧化还原反应。值得注意的是电活性物种在固酶复合物修饰电极界面上仍然可以较为可逆地进行氧化还原反应,考虑到纳米复合物表面基本被酶分子所覆盖(图4C),这一结果暗示纳米复合物表面覆盖的酶分子至少有部分可以视作电极表面-电活性物种之间的“电子介体”。从EIS谱图也可以看出,纳米复合物修饰电极的电阻相对于裸GC电极,明显降低,而固酶纳米复合物的电阻虽然相对于未固酶纳米复合物修饰电极有明显的增加(原因如前所述),但值得注意的是其传荷电阻与裸GC电极相近(与前述分析所得结论相一致)。这一结果表明定向覆盖在纳米复合物表面,充当电子介体的酶分子并没有显著降低电极表面的导电性能,这一结论与文献[6]报道结果有明显不同。
图6A是处于静止状态的Lac/PAn-CoC2O4/GC在N2饱和的PBS(pH=4.4)中扫描获得的CV曲线,附图是处于静态的BOD/PAn-CoC2O4/GC分别在N2,空气,O2饱和的PBS(pH=4.4)中扫描获得的CV曲线。从图6A可以看出:与文献[24,27]报道的固定Lac电极不同,在扫描电位区间内:PAn-CoC2O4纳米复合物修饰电极出现了一对明显的氧化还原峰(基底电极GC在相同电位扫描区间内没有任何与背景电流区分的电化学信号),氧化峰和还原峰电位分别位于431和220 mV附近,还原峰电流明显远高于氧化峰电流(ip,a/ip,c=0.55),此结果与文献[16]报道的聚苯胺接枝功能化多壁碳管修饰电极类似(氧化还原峰电位差接近300 mV),但氧化还原峰电位差明显减小(210 mV),参照文献[16-17]所得结论,这对可逆性较差的氧化还原峰可归于N,N′-二苯基联苯胺阳离子自由基-N,N′-二苯基联苯胺电对发生的氧化还原反应(得失电子数n为2)。如前所述,由于Co-N之间的相互作用对该氧化还原反应产生了较大的影响,因此氧化还原峰整体负移了将近340~410 mV;与之类似,固酶电极Lac/PAn-CoC2O4/GC在相同电位扫描区间范围内也出现了一对位置相近的氧化还原峰(氧化峰和还原峰分别位于439和269 mV,氧化还原峰电位差缩小了40 mV),氧化还原反应可逆程度略有提高(ip,a/ip,c=0.61),其中值电位354.0 mV vs NHE,远离Lac活性中心T1或T3的式电位区间(文献[28]报道700~800 mV vs NHE),而更接近Lac活性中心T2的式电位区间(文献[28]报道约为400 mV vs NHE)。值得注意的是,该电极在固酶前后氧化还原峰面积从1.95×10-6A·V减少到1.35×10-6A·V,鉴于固酶纳米复合物修饰电极氧化还原峰的出峰位置和峰面积与未固酶电极存在差异,可以合理推测这个减弱的氧化还原峰是电极表面固载酶活性中心T2与纳米复合物含有的电活性基团单独或联合作用的得失电子过程所致。但如果要确定这个电化学信号具体是酶活性中心单独产生还是酶活性中心和纳米复合物中所含电活性基团共同产生,还需要更多证据来作出判断。

图6 (A)处于静态的Lac/PAn-CoC2O4/GC和PAn-CoC2O4/GC分别在N2饱和的PBS(pH=4.4)中以5 mV·s-1扫描获得的CV曲线,附图是静止的BOD/PAn-CoC2O4/GC分别在N2,空气和O2饱和的PBS(pH=4.4)中以50 mV·s-1扫速扫描所得CV曲线;(B)静止的Lac/PAn-CoC2O4/GC在不含电子介体的无氧PBS(pH=4.4)中以不同扫描速率得到的CV曲线,附图为对应的阴极,阳极峰电流与扫描速率平方根关系拟合曲线;(C)静止的Lac/PAn-CoC2O4/GC在N2饱和的PBS (pH=4.4)中以20 mV·s-1扫描,不同脉冲高度下获得的DPV曲线Fig.6(A)CV curves of Lac/PAn-CoC2O4/GC and PAn-CoC2O4/GC in N2saturated PBS(pH=4.4)recorded at scan rate:5 mVs-1, respectively,electrodes in static mode,inset:CV curves of BOD/PAn-CoC2O4/GC recorded at scan rate:50 mV·s-1in N2,air and O2saturated PBS(pH=4.4);(B)CV curves of Lac/PAn-CoC2O4/GC electrode in deaerated PBS(pH=4.4)in the absence of electron mediator recorded at variable scan rates of 2,5,10,20 and 50 mVs-1when electrode in static status,inset:fitting plots of corresponding cathodic,anodic peak currents versus square root of scan rates; (C)DPVs of static Lac/PAn-CoC2O4/GC recorded at 20 mV·s-1in N2saturated PBS(pH=4.4)with variable pulse height, from curve a to j corresponding to pulse height at 10,20,30,50,100,200,300,400,500,600 mV
证明出现的电化学信号是属于电极表面固定Lac活性中心与导电基体间直接电子迁移的证据包括:固载其他氧化还原酶的纳米复合物修饰电极上能观察到不同于固定Lac纳米复合物修饰电极CV曲线上的氧化还原信号,这表明此种纳米复合物吸附其他的氧化还原酶后,得到的修饰电极也可以实现酶-电极间的直接电子迁移,但随着酶-载体间相互作用和电子迁移机制的不同,表现出不同的电极反应动力学机制(限于篇幅,仅以与Lac结构和催化活性相近的BOD为例,参见图6A附图)。从图6A附图可看出,固定BOD的纳米复合物修饰电极在N2饱和的电解液中扫描所得的CV曲线上,除了出现上述纳米复合物中电活性基团氧化还原反应的微弱电信号之外,还分别于690和613 mV vs NHE出现了较弱的氧化和还原峰(ip,a/ip,c=1.55,反应可逆性明显优于固定Lac的纳米复合物修饰电极),其中值电位为651.5 mV vs NHE,更接近BOD活性中心T3或T1的式电位[28]。根据纳米复合物修饰电极固定Lac前后的CV曲线上氧化还原峰面积变化(图6A),固酶前后电极活性表面积的变化以及电位扫描速率等参数,依据文献[24]方法可估算出粗略估算出导电酶分子的表面浓度下限为4.2×10-8mol·cm-2。此数值明显高于文献报道[24,27]的纳米多孔金和树枝型聚合物/导电聚合物修饰电极表面的导电酶分子浓度(分别为2.1×10-11mol·cm-2和3.8×10-10mol·cm-2)。而根据电极表面固定漆酶的量(由石墨炉原子吸收法测定得到载体对酶的担载量、漆酶平均分子量、固酶载体活性表面积以及电极表面修饰的固酶复合物质量可以求算)4.0×10-9mol,可以估算出导电酶分子占载体总固载酶分子的70%,这表明依靠部分酶分子在固酶载体表面的特定取向吸附,可以大大提高载体表面固定酶分子实现直接电子迁移的几率,这一结论与前文[29]报道的聚芳酰胺包覆多壁碳纳米管固定Lac修饰电极测试结果相一致。此外值得指出的是,无论是使用CTS作为载体固载Lac还是单独使用CTS作为电极表面修饰剂,都无法实现酶-电极间的直接电子迁移,即该固酶电极在不含底物的溶液中既不能观察到任何与背景电流区分的电化学信号,也观察不到有效催化氧还原的信号,因此CTS仅仅作为成膜剂使用,对酶-电极间的有效电子迁移和电催化反应没有有效贡献。
图6B和附图更进一步给出了该纳米复合物固定Lac电极在相同电解液中以不同电位扫描速率获得的CV曲线以及对应阴阳极峰电流与扫描速率关系拟合曲线,从图6B及其附图可以看出:阴阳极峰电流均随着电位扫描速率的升高而增加,而且在测试速率范围内均与扫速平方根保持良好的线性关系,阴阳极峰电流之比值ip,a/ip,c随着扫速的提高从0.22升高至0.60,而阴阳极峰电位分别负向和正向移动,阴极峰电位负移程度明显高于阳极峰电位(这暗示随着扫速上升,阳极电化学氧化速率明显升高,即高扫速时酶促N,N′-二苯基联苯胺氧化反应受到抑制)。上述结果表明该电极表面固载的Lac活性中心T2与导电基体间发生的直接电子迁移过程为薄层控制型准可逆氧化还原反应,可以理解为纳米复合物中电活性基团通过所谓“摆动”的模式实现固定的酶活性中心-纳米复合物中电活性基团间的电子传递,这与文献[30]报道的固定BOD的氧化还原水凝胶修饰电极的电子迁移机制类似。从图6B附图给出的阴阳极峰电流-扫速平方根关系曲线拟合所得的斜率,结合前述电极反应得失电子数n,电极表面电活性物种浓度以及电极活性表面积等参数,按照文献[31]给出方法可以估算出电极表面薄层中得失电子过程的电对“摆动”引起的表观扩散系数D,为1.02×10-7cm2·s-1。利用文献[23]提及的Nicholson公式,根据求算的表观扩散系数D,峰电位差的依赖参数Ψ,电位扫描速率v,实验温度T以及电极反应得失电子数n等参数,则可以估算出该固酶纳米复合物修饰电极上异相电子迁移速率khet为5.6×10-5cm·s-1,再利用文献[32]给出的电极表面反应层的定义和反应层厚度计算公式Δ=DKM/(kcatC),结合电活性物种表观扩散系数D,电极表面酶分子浓度C,酶催化电活性物种反应速率[32]kcat以及酶对电活性物种的米氏常数[16-17]KM就可以估算出反应层厚度为0.04 cm,以估算的khet对求算的反应层厚度进行归一化处理后所得电极表面异相电子迁移标准速率常数为0.017 s-1。
图6C为纳米复合物固定Lac电极在无氧的PBS(pH=4.4)中以不同的脉冲高度,相同的电位扫描速率获得的DPV曲线,从图6C可以看出当脉冲高度较小时,曲线上的氧化峰电位与图6A的CV曲线上的氧化峰电位接近,而当脉冲高度较大时,DPV曲线上的氧化峰电位逐渐负移到接近氧化还原电对N,N′-二苯基联苯胺阳离子自由基-N,N′-二苯基联苯胺的中值电位的位置,这暗示后者得失电子的电化学活性要高于实现直接电子迁移的酶活性中心。值得注意的是,和文献[33]报道的结果不同,此固酶纳米复合物电极在扫描电位区间范围内只出现了一个单独的氧化峰,并没有观察到两个式电位接近的电活性物种发生电子转移时产生的信号重叠现象,结合前述的出峰电位数值,可以合理推测当脉冲高度较小时固酶活性中心的T2铜离子作为首要电子给体,释放出电子给导电基体,而当脉冲高度较大时,酶活性中心仅作为电子介体,观察到的电化学信号可归因于纳米复合物中电活性基团释放电子的氧化反应。
2.2.2固酶纳米复合物修饰电极的催化氧还原(ORR)性能
图7是处于静态的Lac/PAn-CoC2O4/GC分别在氧气饱和和无氧的不含任何介体的PBS(pH=4.4)中扫描获得的CV曲线。从图7可以看出,固酶电极在氧气饱和以及无氧PBS中的CV曲线存在明显差异:尽管两者正向扫描所得的氧化峰峰电位和峰面积基本保持不变,但是固酶电极在氧气饱和溶液中负向扫描时,于460 mV处还原电流急剧增加,直到~220 mV时达到第一个极限电流(这可以归因为电极表面固载Lac化学氧化N,N′-二苯基联苯胺产物将电子传递给电极表面固定Lac吸附的氧分子,使之电化学还原的过程),随着电位继续负移,电流进一步迅速增加直到~-260 mV处达到第二个极限电流(这可以归因于固酶纳米复合物化学吸附的氧分子在超电势较大时电化学还原的过程,一个旁证是将固酶复合物修饰电极放入空气饱和的电解液中过夜后,结果显示溶液中氧气浓度降低到初始值的大约78%),这充分表明电极表面固定的酶有效催化了氧还原,这一现象与图6A附图中给出的固定BOD纳米复合物在相同电解液中测试结果相一致,不再赘述。值得注意的是未固酶电极PAn-CoC2O4/ GC没有明显的催化氧还原效能,即该电极在无氧和氧气饱和的溶液中扫描所得的CV没有明显区别。相对于氧在此条件下还原的平衡电位970 mV vs NHE,固酶电极催化氧还原的超电势虽然高达510 mV,但仍然比类似电子迁移机制的固定Lac电极[27,34](前者在相同电位扫描区间范围内没有明显的催化氧还原效能,后者氧还原起始电位为400 mV vs NHE)的氧还原超电势小得多,与文献报道固酶电极[24]实现直接电子迁移时的氧还原起始电位(530 mV vs NHE)接近。与文献[6]报道的固酶电极不同的是氧化峰电流没有明显下降,这暗示虽然电极表面处于氧化态的Lac活性中心T2接受了氧气给予的电子被消耗,但同时又将电子传递给了氧化态的N, N′-二苯基联苯胺阳离子自由基,从而使氧化峰电流相对保持恒定。以获得的稳态催化还原电流(以200 mV时为准)对电极活性面积归一化就可以得到稳态催化还原电流密度j为4.2μA·cm-2,结合前述估算的导电固酶分子的表面浓度Γ,由公式:j=nkcΓF(kc为表观电催化氧还原反应速率常数,n为氧还原反应得失电子数,一般认为电极表面固定的Lac无论是通过电子介体介导,还是通过直接电子迁移的方式,氧还原都是按照四电子机制进行,氧直接被还原为水而不会形成中间体过氧化氢。文献[30]也指出以极限扩散电流对旋转圆盘电极旋转角速率平方根作图,根据拟合曲线所得斜率就可估算出氧还原反应得失电子数为4)便可以估算出此电极上固定Lac催化氧还原为水分子的表观速率常数上限为2.6×10-4s-1,这一数值明显低于文献[35]报道的数值(0.07 s-1),与吸附Lac的碳纳米管修饰电极[36]接近(根据数据估算为2.2×10-4s-1),这一数值接近于Lac活性位T3上形成的配合物分解为水分子的反应速率[28](0.034 s-1),即电催化氧还原的过程实际上受制于酶分子活性位上吸附氧形成的配合物分解的过程。这暗示固酶纳米复合物与固定Lac分子之间的复杂相互作用对固定Lac的催化效率存在较大影响,这与PAn-CoC2O4特殊的化学结构和表面形貌有密切关联。

图7 Lac/PAn-CoC2O4/GC分别在不含介体的为O2和N2饱和的静止PBS(pH=4.4)中以2 mV·s-1的扫速扫描所得CV曲线Fig.7 CV curves of static Lac/PAn-CoC2O4/GC electrode recorded at rate:2 mV·s-1in N2bubbled and oxygen-saturated PBS(pH=4.4)in the absence of external mediator,respectively
2.2.3固酶电极催化性能的重现性、长期使用性、热稳定性及pH值耐受性
图8A为同一批次制备的固酶电极置于氧气饱和的PBS(pH=4.4)中扫描所得CV曲线上极限催化电流密度的对比图,从图8A可以看到5个按同一工序制备的Lac/PAn-CoC2O4/GC电极极限催化电流密度显著性差异较小,这表明该固酶电极催化氧还原性能具有良好的重现性;图8B为Lac/PAn-CoC2O4/GC于氧气饱和的PBS中扫描所得极限催化电流密度与电极低温储存时间的关系曲线。从图8B可看出:新制备的Lac/PAn-CoC2O4/GC电极在氧气饱和的PBS中立即进行测试和此电极在4℃冰箱中储存7 d后测试得到的极限催化电流差异很小(极限催化电流密度的降低幅度仅为10%),而且氧还原起始电位没有明显改变(通过对比两者的CV可以看出,图不再给出),随着储存时间延长固酶电极催化氧还原性能随之缓慢下降,当低温储存时间超过10 d后电极催化氧还原性能下降迅速,电极储存21 d后催化性能仍然可以保留初始值的大约47.2%,这一结果要优于文献[37]报道结果(同样温度下储存21 d后性能下降到不足初始值的15%)。图8C为固酶电极在氧气饱和的不同pH值PBS溶液中以相同扫速(2 mV·s-1),常温下测定的稳态催化电流密度-pH关系曲线,以及最佳pH值条件,相同扫速(2 mV·s-1)下测定的极限催化电流密度-温度关系曲线。从图8C可以看出:纳米复合物固定Lac修饰电极催化氧还原性能与游离Lac类似,存在一个最佳pH值,但与游离Lac不同,最佳pH值出现在4.4,这表明载体表面固定的Lac由于催化活性中心所处的微环境发生了明显的改变,进而使得催化活力-pH关系也与游离Lac存在明显差异,据此pH= 4.4被设定为测试固酶电极催化氧还原性能的工作pH值;此外还可以观察到在测试温度范围内,随着温度升高,在328 K之前极限催化电流密度随温度线性升高,而当温度超过328 K之后极限催化电流密度迅速下降,这个结果与文献[30]报道固酶电极的催化电流密度-温度关系非常相似,这表明此电极表面的固酶载体很可能以化学吸附的形式固定Lac,而当温度升高后酶变性从而造成电池输出性能下降(温度升高后并没有检测到酶分子从电极表面的固酶载体上脱落)。根据图中数据可以估算出电池催化反应的表观活化能大约是43.5 kJmol-1,而酶变性的活化能则大约是66.3 kJ·mol-1。
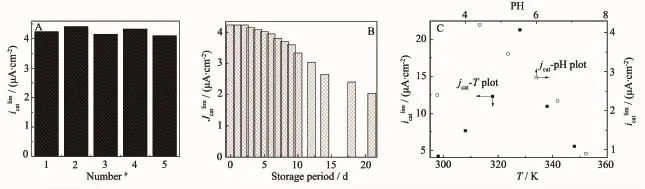
图8 Lac/PAn-CoC2O4/GC催化氧还原性能的重现性(A),长期使用性(B)以及温度/pH-催化性能间的依赖关系(C)Fig.8 Reproducibility of catalysis for ORR(A),long-term stability of catalysis for ORR(B)and dependence of its catalytic effect on temperature or pH(C)for Lac/PAn-CoC2O4/GC

图9 计时电流法表征Lac/PAn-CoC2O4/GC对氧传感的性能,插图为对应的极限催化电流密度-氧气浓度双倒数关系曲线Fig.9 Performance for oxygen detection of Lac/ PAn-CoC2O4/GC characterized by chronoamperometry in stirring PBS(pH=4.4), electrode in stationary status and applied potential at 160 mV,inset is corresponding double reciprocal plot of limited catalytic current density vs concentration of substrate-dioxygen
2.3固酶电极作为氧电化学传感器的性能评估
漆酶除了能化学催化氧化特定结构的酚类化合物和一些杂环化合物之外,还能够同时按照四电子机制电还原化学吸附的氧分子为水。一般认为只有当酶-电极间可以实现有效的电子迁移(无论是通过使用电子介体还是以直接电子迁移的方式)时,才能够通过检测响应电流的方式以确定酶催化反应底物的氧分子的浓度[6,29]。图9为Lac/CoC2O4/GC电极在不含电子介体的无氧PBS(pH=4.4)中,分批加入不同体积空气饱和的PBS(pH=4.4)时产生的响应电流密度与时间关系曲线,插图则是固酶电极的稳态催化电流密度倒数与底物氧分子浓度倒数间的关系校正曲线。从图9及插图可以看出:固酶电极可以有效催化氧分子还原,对其响应迅速。此电极在氧气浓度0.43~7.5μmol·L-1的范围内,稳态催化电流与氧气浓度均能保持良好的线性关系。这一固酶电极对氧的灵敏度为0.006μA·L·μmol-1,这一数值虽远低于文献[6]报道的壳聚糖-多壁碳纳米管固定Lac电极对氧分子的灵敏度(27.3μA·L·μmol-1),但其对氧的检测限(检测限的定义是背景电流平均值加上3倍背景信号标准偏差所得的电流和,在响应电流-底物浓度工作曲线上对应的浓度值)可以低至0.20μmol·L-1,这一数值仅为文献[6]报道的固酶电极(7.8μmol·L-1)的1/39。从双倒数L-B曲线拟合所得的固酶电极对氧分子的KM数值(122.4μmol·L-1)来看,本文制备的固定Lac的PAn-CoC2O4纳米复合物修饰电极对氧气的亲和力很高,不但远远高于文献[6]报道的固酶电极对氧的亲和力(KM=3.22 mmol·L-1),而且亦高于文献[38]报道的固酶电极对氧分子的亲和力(191.0μmol·L-1)。此外由溶液中氧气浓度-固酶电极浸泡时间关系曲线(图略)的斜率求算出氧分子平均消耗速率V(0.024 5μmol·L-1·s-1),以及电极表面纳米复合物修饰层中固酶物质的量nE(2.8×10-9mol),根据文献[24]给出公式V=kEnE(kE为酶化学吸附氧分子表观速率常数)可以估算kE为0.26 s-1。
将所有获得的酶催化反应相关过程的动力学参数在同一量纲下进行对比发现,固定Lac电化学还原氧分子的速率远低于酶化学吸附氧的速率,也低于酶-导电基体之间直接电子迁移速率,这表明降低吸附的氧分子转化的活化能才是改善这型固酶电极性能的关键,在保留该纳米复合物对氧较高亲和力的前提下,合理调控纳米复合物官能团对氧的结合能力,将可以提升该固酶电极的催化效率。
3 结论
利用PAn-CoC2O4纳米复合物作为固定Lac的载体,依靠化学吸附,物理吸附以及配位作用的协同效应稳定的固载了Lac,进一步制备了固酶基电极。采用光谱学技术手段表征了固酶纳米复合物的结构和形貌,以电化学方法测试了此电极的导电性能,研究了电极表面固酶的直接电子迁移性能及催化氧还原性能,在此基础上评估了其作为氧电化学传感器的性能。实验结果表明:固酶纳米复合物修饰电极在测试电位区间内出现一个式电位接近酶活性中心T2的准可逆电化学信号,具有较快的电子迁移速率,该固酶电极具有较高的氧还原起始电位(460 mV vs NHE)和较快的化学吸附氧分子速率,但酶电催化氧分子还原反应受限于吸附氧分子转化为水的过程。这种纳米复合物固酶电极还显示出较为良好的催化性能重现性,长期使用性,热稳定性以及pH耐受性。该电极作为电流型氧电化学传感器的测试结果表明其具有极低的检测限以及对底物较高的亲和力等优势。
[1]Zheng W,Ma J Y,Guo F,et al.Bio-Med.Mater.Eng.,2014, 24(1):229-235
[2]Fokina O,Eipper J,Winandy L,et al.Bioresour.Technol., 2015,175:445-453
[3]Zhang L L,Bai L,Xu M,etal.Nano Energy,2015,11:48-55
[4]Pankratov D V,Zeifman Y S,Morozova O V,et al. Electroanalysis,2013,25(5):1143-1149
[5]Scherbahn V,Putze MT,DietzelB,etal.Biosens.Bioelectron., 2014,61(21):631-638
[6]Liu Y,Qu X H,Guo H W,et al.Biosens.Bioelectron.,2006, 21(12):2195-2201
[7]Rasmussen M,Ritzmann R E,Lee I,et al.J.Am.Chem. Soc.,2012,134(3):1458-1460
[8]Ivanov I,Vidakovic-koch T,Sundmacher K J.Electroanal. Chem.,2013,690(2):68-73
[9]Zelechowska K,Stolarczyk K,Lyp D,et al.Biocyber. Biomed.Eng.,2013,33(4):235-245
[10]Cardoso F P,Neto S A,Fenga P G,et al.Electrochim.Acta, 2013,90(5):90-94
[11]Farneth W E,Diner B A,Gierke T D,et al.Electroanal. Chem.,2005,581(2):190-196
[12]Zhang L,Zhang Q,Lu X B,et al.Biosens.Bioelectron., 2007,23(1):102-106
[13]Lu X B,Wen Z H,Li J H.Biomaterials,2006,27(33):5740-5747
[14]Royo B,Sosna M,Asensio A C,et al.J.Electroanal.Chem., 2013,704(9):67-74
[15]Filip J,Tkac J.Bioelectrochemistry,2014,96(2):14-20
[16]Manesh K M,Santhosh P,Gopalan A I,etal.Electroanalysis, 2006,18(16):1564-1571
[17]Santhosh P,Manesh K M,Lee K P,et al.Electroanalysis, 2006,18(9):894-903
[18]CAO Zheng-Yan(曹正艳),YANG Chun-Ming(杨春明), LI Hai-Yin(李海银),et al.Chinese J.Appl.Chem.(应用化学),2009,26(11):1264-1268
[19]Blandford C F,Heather R S,Armstrong F A.Chem.Commun., 2007,43(17):1710-1712
[20]HUANG Jun(黄俊),ZHOU Ju-Ying(周菊英),XIAO Hai-Yan(肖海燕),et al.Acta Chim.Sinica(化学学报), 2005,63(14):1343-1347
[21]ZENG Han(曾涵),ZHAO Shu-Xian(赵淑贤),GONG Lan-Xin(龚兰新),et al.Chinese J.Appl.Chem.(应用化学), 2013,30(4):436-443
[22]Zhao H Y,Zhou H M,Zhang J X,et al.Biosens.Bioelectron., 2009,25:463-468
[23]Shleev S,Christenson A,Serezhenkov V,et al.Biochem.J., 2005,385:745-754
[24]Qiu H J,Xu C X,Huang X R,etal.J.Phys.Chem.C,2009, 113(6):2521-2525
[25]Zhu Y F,Kaskel S,Shi J L,et al.Chem.Mater.,2007,19: 6408-6413
[26]ZENG Han(曾涵),YANG Yang(杨阳),LI Xiao-Juan(李小娟),et al.Chin.J.Anal.Chem.(分析化学),2015,43(12): 1794-1800
[27]Rahman M A,Noh H B,Shim Y B.Anal.Chem.,2008,80: 8020-8027
[28]Osman M H,Shah A A,Walsh F C.Biosens.Bioelectron., 2011,26:3087-3102
[29]ZENG Han(曾涵),LIAO Ling-Wen(廖铃文),LI Ming-Fang (李明芳),et al.Acta Phys.-Chim.Sin.(物理化学学报), 2010,26(12):3217-3224
[30]Mano N,Kim H H,Zhang Y C,et al.J.Am.Chem.Soc., 2002,124(22):6480-6486
[31]Mao F,Mano N,Heller A.J.Am.Chem.Soc.,2003,125 (16):4951-4957
[32]Kano K,Ikeda T.Anal.Sci.,2000,16(10):1013-1021
[33]Katz E,Heleg-Shabtai V,Willner I,et al.Angew.Chem. Int.Ed.,1998,37(23):3253-3256
[34]Pita M,Shleev S,Ruzgas T,et al.Electrochem.Commun., 2006,8(5):747-753
[35]Ramasamy R P,Luckarift H R,Ivnitski D M,et al.Chem. Commun.,2010,46(33):6045-6047
[36]Zheng W,Zhou H M,Zheng Y F,et al.Chem.Phys.Lett., 2008,457(4/5/6):381-385
[37]Soukharev V,Mano N,Heller A.J.Am.Chem.Soc.,2004, 126(27):8368-8369
[38]Farneth W E,DAmore M B.J.Electroanal.Chem.,2005, 581(2):197-205
Direct Electrochemistry of Electrode Modified with Thin Film of Laccase Immobilized in Nano-Composite of Polyaniline-CoC2O4
YANG Yang HUO Wen-Shan ZHOU Zheng ZHANG Qi ZENG Han*
(Laboratory of New Energy Materials Chemistry,Chemistry and Chemical Engineering Academy,XinJiang Normal University, Urumuqi 830054,China)
Electrochemical methods including cyclic voltammetry,differential pulse voltammetry,electrochemical impedance spectrometry and chronoamperometry,together with auxiliary means such as F-T infrared spectrometry, Ultra-violetvisible spectrometry,atomic force microscopy,transmission electron microscopy and atomic adsorption spectrometry were used to characterize the chemical composition,structure and morphology of polyaniline-CoC2O4nano-composite,to measure the change of conductivity in nano-composite before and after Laccase immobilization and to investigate the direct electrochemistry of redox protein molecules entrapped in the matrix.Its catalytic effect on oxygen reduction reaction and performance as electrochemical sensor for oxygen detection were evaluated subsequently.Results from tests indicated this Laccase-based electrode shuttled electrons from enzyme active site T2as primary electron acceptor to dioxygen molecules attached chemically in the matrix,achieving the electro-reduction of O2in the absence of any external mediator with its apparent electron transferring rate:0.017 s-1.This laccase-based electrode displayed favorable catalytic effecton oxygen reduction reaction(onsetpotential for catalysis:460 mV vs NHE,apparent turn-over frequency for oxygen reduction reaction:2.6×10-4s-1).Enzymatic oxygen electro-reduction into water should be ascribed to the key process to promote the performance of biocathode.This Laccase based electrochemical sensor had such advantages as extremely low detection limit (0.20μmol·L-1)to oxygen monitoring,wide linear responding range of concentration(0.4~7.5μmol·L-1)and high affinity towards substrate(KM=122.4μmol·L-1).
Polyaniline-CoC2O4nano-composite;Laccase;Direct electrochemistry;Catalytic oxygen reduction reaction;Electrochemical sensor
O643.3
A
1001-4861(2016)12-2117-12
10.11862/CJIC.2016.277
2016-06-13。收修改稿日期:2016-10-14。
国家自然科学基金(No.21363024,31560249)、新疆师范大学博士科研启动基金(No.XJNUBS1228)、新疆维吾尔自治区2013年度高校科研计划青年教师培育项目(No.XJEDU2013S29)、新疆师范大学研究生科技创新项目基金(No.XSY201502009)和新疆师范大学大学生创新创业项目(No.201510762074)资助。
*通信联系人。E-mail:zenghan1289@163.com;Tel:+86-991-4332279
